I hope I never get gout. We see someone with this problem maybe once a month on our admissions unit. The usual story is a middle-aged or elderly person with a fairly sudden onset of severe joint pain. Usually gout can be managed at home, but last week we admitted George, a 68-year-old man living on his own. He had managed to look after himself reasonably well since his wife died last year, but now could not walk because of the excruciating pain in the big toe of his left foot.
Before he came in, George had been fairly well apart from mildly raised blood pressure, for which he took a tablet of bendroflumethazide – a thiazide diuretic.
On the day of admission he had been hopping around the bathroom, slipped and fell against the bath, breaking his arm. As he was describing this I looked at the junior doctor who was with me – she was almost giggling. I gave her a stern look. This conjured up a comical scene, but George did not find it funny.
George had managed to crawl slowly and painfully to the bedroom and call the ambulance. In the emergency department he had complained much more about the pain in his foot than his broken arm. He was very reluctant to let us look–
“don’t touch it – please”.
He had podagra, a wonderful word for gout affecting the first metatarsal-phalangeal joint (the one where bunions appear). For years I’ve been wanting to use the word podagra in Scrabble but not yet got the right letters.
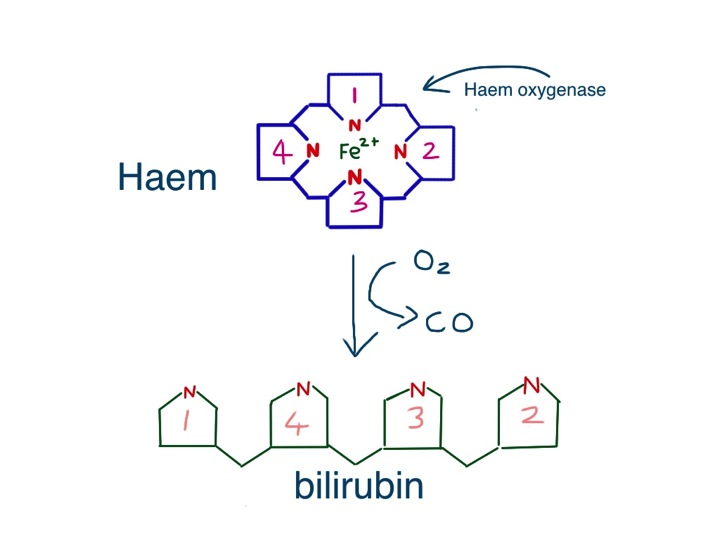
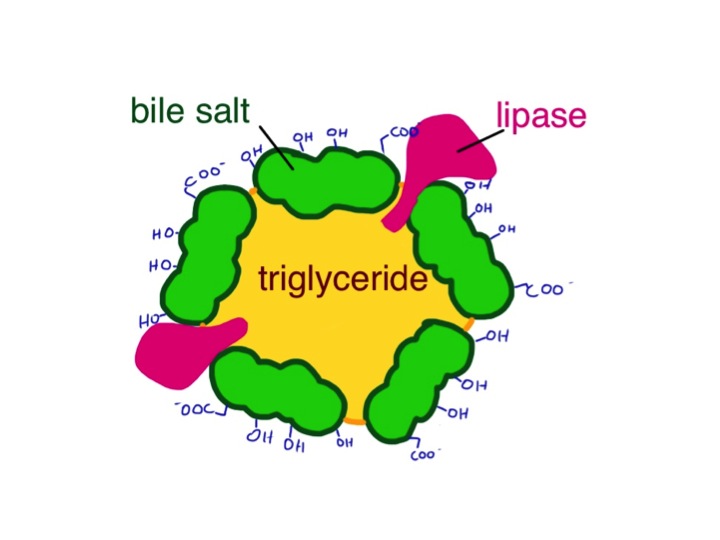
George’s big toe was swollen and red – we did not need to examine it to see how tender it was. The inflammation was caused by the usual culprits – neutrophils. They had been summoned not by germs or allergens, but in this case by crystals of uric acid deposited in the toe joint causing damage to the synovial membrane.
Uric acid is made from purines. You may remember from high school biology that DNA is principally made from four bases A,T,C and G. Cytosine and thymine are pyrimidines, while adenine and guanine are the bigger purines.
the purines adenine and guanine are converted into xanthine and then uric acid which is excreted in urine
A lot of the food we eat contains DNA, whether we are meat-eaters or vegetarians, so we continually have to get rid of the purines. Our bodies also can make the DNA bases from simpler molecules, and unless we are growing or pregnant, we have to get rid of those too.
In humans purines are broken down to xanthine (pronounced zantheen) and then, via the enzyme xanthine oxidase to uric acid. Most other animals, apart from primates, take it further and turn uric acid into allantoin via the enzyme uricase. Allantoin is more soluble and more easily excreted. It is not clear why evolution has decided that we humans do not need this final step, which would make sure we never get gout. There has been the suggestion that uric acid is an important antioxidant which helps protect us – more recently others have suggested that uric acid, or urate is damaging.
What is clear is that fructose is important. Fructose is a sugar which is found in fruit juice and honey – sounds very healthy. But there is now increasing evidence that fructose is bad for us, at least in the large quantities we are now consuming.
Table sugar or sucrose is half fructose. It is a di-saccharide, one molecule of glucose and one of fructose joined together. Fructose is not metabolised in the same way as glucose.
sucrose is a disaccharide of glucose and fructose
It is converted straight away to fat – triglycerides. The pathway involves the addition of a phosphate group to fructose by the enzyme fructokinase. This uses adenosine triphosphate (ATP), and it seems that the adenosine used in this pathway is then broken down and converted, via xanthine oxidase, to uric acid.
Fizzy drinks contain a lot of sucrose
George used to drink at least a glass of sweetened orange juice and two or three cans of coca-cola every day. This ensured he had plenty of fructose which increased his uric acid production, and the thiazide diuretic reduced his kidneys’ ability to excrete it.
As well as increasing uric acid production, fructose seems to promote the formation of intra-abdominal fat. If Robin Lustig is to be believed, fructose, derived from sucrose and high fructose corn-syrup, is much more important than saturated fat in causing obesity, diabetes and vascular disease. His videos and book are worth looking at:
What really interests me is that milk contains loads of the enzyme xanthine oxidase – the enzyme which converts xanthine into uric acid. Much more than anywhere else in the body. Why on earth would you need this enzyme which converts xanthine to uric acid in milk? It seems that this enzyme can do other things apart from oxidising xanthine, it can also generate some of the toxic compounds such as superoxide which we previously saw being used by neutrophils to kill bacteria.
milk contains lots of xanthine oxidase
Now this is just a theory – but if you happen to be a research microbiologist maybe you can test it. Milk is a suspension of fat globules in a protein-rich watery soup. But not ordinary fat globules. Each tiny blob of fat in milk is covered by a a complex phospholipid trilayer in which is embedded lots of xanthine oxidase enzyme.
The theory I’d like to put forward is that nasty bacteria like salmonella, which cause gastroenteritis by invading and attacking the cells which line our intestine, will also attack milk fat globules. Milk globules have some of the same proteins in their outer membrane as intestine cells.
cartoon of a section of a milk globule showing three layers of phospholipid membrane, surface proteins and xanthine oxidase
When they attack they activate the xanthine oxidase enzyme which generates superoxide which will eventually kill them. Non-invasive, non-agressive germs which we want to colonise our guts such as commensal E.coli and lactobacilli would not activate the enzyme and be allowed to survive. It is only an hypothesis at the moment, but would seem to be a great way to make sure that new-born infants, when fed on breast milk with lots of xanthine oxidase, get the right germs in their guts. We know that babies fed on formula, which contains less xanthine oxidase are more likely to get problems with diarrhoea.
It would also explain why yoghurt, which is made by deliberately allowing germs to breed in it, only very rarely contains nasty germs such as salmonella (although there is the notorious case of a batch of hazlenut yoghurt which had botulinum toxin – clostridium botulinum causes problems by synthesising a toxin, not by physically attacking cells).
All cancer chemotherapy is unpleasant, but the chemo we use for breast cancer is particularly nasty. Megan is 48 and was admitted this week with a fever and diarrhoea, a week after her third course of CEF – cyclophosphamide, epirubicin and 5-flurouracil. Her breast cancer was diagnosed a few months ago – she noticed a lump in her left breast when washing herself in the bath. Megan could tell from the face of her GP when she was examined that it was going to be bad.
Then, shortly after, there was the ultrasound, mammogram and trucut needle biopsy at the breast clinic which hurt more than she was expecting – Megan was really scared by this time.
mammogram of breast showing a cancer bottom right
She remembers vividly the meeting with the breast surgeon a week later, but only as a jumble of “surgery”, “partial mastectomy”, “total mastectomy”, “node clearance”, “chemotherapy”, “radiotherapy”. Her husband Jack was with her, but it didn’t make much sense to him either. They went home with a pile of leaflets and phone numbers.
The surgery went fine, everyone was really nice and caring. Then a meeting with the oncologist. She advised chemotherapy then radiotherapy. That seemed much more simple. But chemotherapy was awful. Megan felt sick and hopeless. All she could do was sit in bed and do crosswords. Just as she was starting to feel better the next round of chemo was due.
epirubicin is an anthracycline antibiotic chemotherapy agent which works by damaging DNA
When I met her she had the look of a usually bright and cheerful person who was wondering if she could take any more. I told her that her white cell count was low, and we needed to give her intravenous antibiotics until her fever resolved and white cell count recovered.
How does cancer chemotherapy work? Why does it kill cancer cells in preference to normal cells in our body?
The normal answer I get from the young doctors and students is that chemotherapy targets rapidly-dividing cells. It really is not quite as simple as that. Breast cancer cells divide much less rapidly than bone marrow or gut epithelial cells, but chemotherapy is designed to kill all the cancer cells and leave our bone marrow and intestinal lining intact. To answer the question properly, we need to understand what makes cancer cells malignant.
We have a much better understanding of the molecular basis of cancer than we did thirty years ago. Cancer cells usually have a number of genetic abnormalities which result in the cells no longer behaving in a useful and regulated way. Cells, whether they are from breast, colon, lung or any other tissue need to behave in a regulated and responsible way. They need to co-operate with the cells around them, working together to make sure the organ works properly. Like in normal society, where people have to abide by rules which make sure everything works properly. If someone misbehaves badly, the police force and courts act to stop them doing the bad thing. Similarly, if a cell is damaged and behaves abnormally, the body has ways of making sure that is either removed or repaired. Our immune system is constantly removing abnormal cells, and all cells have a mechanism to detect and repair DNA damage. One of the main mechanisms to identify DNA damage is the p53 system. The p53 gene is turned on when DNA damage is detected. This causes p53 protein to be made and cause the cell to stop dividing until the DNA damage is repaired. If the DNA cannot be repaired the cell undergoes programmed cell death. Harsh, you may think, but necessary to prevent the cell dividing and making more abnormal cells – “here is a pistol – I think you need to go to the library and consider your options”.
When normal (non-malignant) cells have damaged DNA which cannot be repaired they do the decent thing
The problem comes when the law enforcers are compromised. Corruption in the police force or judges is disastrous for society and for our bodies. Most common cancers have damage to the p53 gene, meaning that cells will divide even when DNA is damaged. Understanding this helps us understand why traditional chemotherapy targets cancer cells.
Cyclophosphamide is a nitrogen mustard alkylating agent which damages DNA by permanently crosslinking the two DNA strands. Nitrogen mustard has been molecule of the month: find out more about its chemisty here:
In normal cells this damage is detected and prompts an activation of the p53 gene causing production of p53 protein – with consequent shutdown of cell division until the damage is repaired (or, if the damage cannot be repaired the cell commits sucicide). In cancer cells which cannot make p53, the cells will continue to divide and render them more susceptible to further DNA damage. This is because when cells are dividing the DNA is unravelled and more exposed to the damaging chemotherapy agent.
Continued division of cancer cells will also make them more susceptible to 5-fluorouracil. This drug inhibits the enzyme thymidylate synthase, preventing the production of normal thymine, one of the DNA bases. The cells are trying to divide but cannot make thymine and suffer a fate known as “thymineless death”. Cells which have normal p53 will have shut down and stopped dividing, and will not suffer in this way.
Epirubicin is a member of a group of drugs called anthracycline antibiotics, derived from steptomyces bacteria. These substances also work by binding to DNA and interfering with cell division, again targeting the breast cancer cells which are still dividing, despite having their DNA damaged, because of defective p53.
This combination of drugs work well to kill the breast cancer cells, but the shutdown of normal bone marrow production and intestinal epithelial cell division still causes a major problem, as evidenced by Megan and her neutropenic sepsis and diarrhoea. Without white cells and gut epithelial cell production she cannot not defend herself against normal germs.
Patients undergoing chemotherapy are given a whole pile of leaflets including advice about neutropenic sepsis
Damage to the p53 gene is not the only abnormality that causes breast and other cancers. Most breast cancer cells have a large number of genetic abnormalities, many of which we know are important in making the cells cancerous, but we do not know how. There is a really good video which explains what we know about the genetic abnormalities in breast cancer and other cancers by Prof Sir Mike Stratton FRS –
Also, it is being recognised that epigenetic abnormalities are common in cancer cells, with differences in DNA methylation between normal and cancer cells. As I mentioned in a previous post, epigenetic changes are likely to be important in ageing, as well as cancer. I think there maybe an important link between these.
What causes the DNA damage in breast cells? That is a big problem to understand. It is easy to imagine how lung epithelial cells are exposed to cigarette smoke which contains carcinogens, and how colon epithelial cells may be damaged by nasty substances in our diet. But breast cells – why do they get damaged? The main risk factors for breast cancer are:
1 early menarche (onset of menstrual periods at an early age
2 late first pregnancy
3 fewer pregnancies
4 obesity
5 alcohol
How do these factors damage DNA and cause cancer? I do not know. Answers please.
Now on to the food link – garlic.
garlic has its characteristic odour because it contains the molecule allicin, which helps protect it from bacteria and insects which want to eat it
Cyclophosphamide is a nitrogen mustard. It is called that because mustard gas was thought to smell like mustard. In fact it smells more like garlic.
poster from first world war – wikimedia commons
Garlic contains a substance known as allicin, which has a similar molecular shape to nitrogen mustard.
allicin and nitrogen mustard have a similar molecular shape – which is maybe why they have a similar smell
It has been suggested that garlic has all manner of wonderful properties to keep us healthy, including lowering cholesterol, preventing heart attacks and protecting us against infection (and vampires). The hard evidence is not there. But as Neils Bohr, the eminent physicist, said when a visiting journalist remarked “ why Professor Bohr, do you have a horseshoe above your front door – surely you cannot believe that it will bring you good luck?” replied:
“Of course I don’t believe in it, but I understand it brings you luck, whether you believe in it or not.”
Sarah was hiding under her bedclothes. She is 24, and has had a really miserable time recently. Last week she was sacked from her underpaid job, which she hated anyway. Her Mother and Ken (her Mother’s new partner) have been putting pressure on her to “get her life sorted out”.
She had seen her GP, who thought she was depressed and organised some group cognitive behaviour therapy. Sarah did not like the group leader, he was too much like Ken. Her GP then gave her some sertraline tablets – she was told they would not work straight away but might take two weeks – that was last week.
She was planning to move into a flat with her new boyfriend, but yesterday found out he was seeing someone else. Life seemed totally hopeless and she drank half a bottle of vodka and took thirty paracetamol tablets.
paracetamol is sold in boxes of only 16 in the UK to reduce the likelihood of overdose
She left the empty packets on the kitchen table – she admitted that she wanted her Mother to find them when she returned from the pub with Ken. They found her in her room, semi-conscious from the vodka, and rang for an ambulance.
In the emergency department she had blood taken, and the paracetamol level was high enough to need treatment. She was started on a drip containing acetylcysteine (Parvolex), about five hours after taking the overdose, and sent to our assessment ward.
December is always a popular month for deliberate self-harm. It might be the “dark days before Christmas”, with insufficient sunshine to activate the pineal gland. It also seems that Christmas puts a lot of tension on families, and makes some people very miserable, rather than happy. My Mother-in-Law remembers Christmas as a child:
“the smell of sprouts and the sound of weeping from the kitchen”
We have medical students on the ward, who have all recently spent time in primary care. I ask
“Do you see a lot of depression in GPs surgeries?”
They smile as they recount the very large numbers of patients who attend their family doctors because they are unhappy and seem unable to cope with their lives.
We look after a lot of people who have taken a deliberate overdose of drugs. Many are young and female, and when we go to see them in the morning, they are often hiding under the bedclothes – reluctant to talk. Sarah was not keen to talk to begin with, but soon told us the whole story. She said she felt really stupid now, and felt she had wasted everyone’s time. She was glad she had not managed to kill herself. We told her that she would soon be seen by our psychiatry colleagues. They sent her home later that day, after we had checked her blood results and were sure she had not done any serious damage to her liver. She has an outpatient appointment with the psychiatric team next week.
Overdose of paracetamol, or acetaminophen as it is called in the US, is the commonest reason for acute liver failure needing liver transplant in the UK and US in young people. Why is it so toxic in overdose?
Paracetamol is metabolised by our liver. Normally, about 90% is conjugated with glucuronic acid or sulphate. Glucuronic acid is like glucose, with an extra COOH attached.
liver metabolic pathways for paracetamol – Fvasconcellos wikicommons
Once conjugated paracetamol is much more soluble in water, and is excreted in the kidneys. But about 10% is metabolised by liver cytochrome enzymes. They are called cytochromes because they make the liver cells coloured – a brownish red colour. Cytochromes enzymes contain haem, but mostly with the iron in the Fe3+ form. This makes them brownish rather than the bright red in haemoglobin and myoglobin, where the iron is in the Fe2+ oxidation state. That is why liver is a different colour from ordinary meat. These cytochrome enzymes are also known as p450 enzymes. This is because if a pure solution of the enzyme in a test tube is bubbled with carbon monoxide, the enzyme absorbs light with a peak at 450nm – purplish blue. Absorbing blue light means that it looks reddish – the colour which is not absorbed. There are a whole range of cytochrome p450 enzymes in the liver – to deal with the vast number of chemicals which our bodies need to deal with. Their job is to oxidise the variety of chemicals in our diet and render them soluble and make it easier for them to be excreted by our kidneys.
The small proportion of paracetamol which is oxidised by p450 enzymes is turned into a toxic chemical – NApQI – this stands for N-acetyl-p-quinone-imine. This molecule is only very slightly different from the parent paracetamol molecule, but contains a C=O group on the benzene ring, making it a quinone. Quinones are often reactive chemicals. They are used industrially to generate hydrogen peroxide. NApQI will rapidly damage the liver cells that make it from paracetamol. But this damage does not happen when we take the normal dose. Fortunately all of the NApQI is rapidly detoxified by reaction with glutathione, a substance normally present in the liver cells, to produce a non-toxic conjugate which is excreted in the urine.
The problem comes when paracetamol is taken in overdose. The glucuronic acid and sulphate conjugation pathways are overwhelmed, and more of the paracetamol is turned into NApQI by cytochrome p450 enzymes. After a short time the glutathione runs out and NApQI cannot be detoxified and causes liver damage. The acetylcysteine we gave to Sarah as soon as she came in is used by liver cells to replenish glutathione and prevent any more liver damage. We give a lot of acetylcysteine – about 25 grammes in total – about an ounce, over a period of 21 hours. After this time all of the paracetamol should be eliminated as non-toxic substances. Chemicals that cause induction of p450 enzymes, such as alcohol and anticonvulsant drugs, can make paracetamol toxicity more likely. Malnutrition, which can deplete glutathione reserves, can also increase the risk of liver damage.
The second question I will ask, but not give a satisfactory answer to, is why does it take a long time for antidepressant drugs like sertraline to work? This drug, like Prozac, or fluoxetine, is thought to increase the amount of serotonin available in the brain. Serotonin is a small molecule, made from the amino acid tryptophan. There is a lot of tryptophan in certain foods such as soyabeans and cheese, but smaller amounts in all protein sources. Sertraline is an SSRI – a selective serotonin reuptake inhibitor. Serotonin is released from one nerve and travels across a synapse to cause an effect of the next nerve. Following release serotonin is taken back up into the cell that released it, causing the effect to terminate. Blockade of this uptake will mean that there is more serotonin available to have its effect. Drugs such as ecstasy (MDMA) are thought to have their euphoric effect by increasing serotonin levels, but they work more or less straight away.
I think we have an awful lot to learn about how the brain works. In fact I’m sure that we have very little idea. We don’t even seem to understand the basic operating system.
The information for this red apple is coded in the silicon chips of your computer as 0s and 1s, in a particular sequence to allow the screen to form this image. How does it work in your brain?
If I ask you to close your eyes and think of an apple, a nice red one, that is easy to do. But what is that thought? The thought – the image of an apple in your mind is certainly a thing – a physical thing. Although you think you are only imagining the picture of the apple – the picture must be there – somewhere. The thought must be made of something – even if it is a perturbation of electrical charge or change in chemicals. And it is certainly in your brain. But if I ask you exactly what the thought is made of, and where in your brain it is, we have no idea. No idea at all. Is the thought of an apple in the shape of an apple? – probably not, but of course it might be.
I have an analogy for how much we understand how the brain works. Let us imagine that you find an Amazonian tribe which has had very little contact with industrialised society. You take them a plasma-screen television, a large solar panel to recharge the batteries, and a satellite dish. You put them together in a clearing in the forest, charge up the battery and turn the television on. Adjustment of the satellite dish results in the face of Simon Cowell presenting the X-Factor appearing on the television.
Simon Cowell photo courtesy Alison Martin of SimonCowellOnline.com.
The tribespeople are intrigued. You leave them with the whole kit. They start experimenting with it – they realise that moving the satellite dish changes the clarity of the image, they realise that they can change the picture or increase the sound volume by pressing various buttons. One enterprising young man has even unscrewed the back of the television and discovered that some components are removable, and taking them out does all sorts of interesting things to the picture and sound. They feel that after a few months they have a pretty good idea about how the television works. That is about the level of understanding we have of how our brains work – we can give people chemicals which change conscious level and behaviour, we can operate and stimulate certain parts of the brain to see what happens, and can use functional MRI to see what part lights up when we do, or think about certain things. But we don’t know how it works. Not a clue.
Now for the food link – grapefruit. We have known for some time that drinking grapefruit juice can cause potentially serious interactions with certain drugs which are metabolized by liver p450 enzymes. This was discovered when researchers, who were looking for an effect of alcohol on the blood pressure drug felodipine, mixed the alcohol with grapefruit juice to disguise the taste. They found that the grapefruit juice, rather than the alcohol, had a huge effect on the rate at which felodipine was metabolized by the liver. This is because it contains a substance which binds to and inhibits certain cytochrome p450 enzymes. Grapefruit also affects the metabolism of many other drugs, such as statins and oral contraceptives – for more information see: http://www.healthcentral.com/peoplespharmacy/pp_guides/pdf/gfruit02.pdf
Robbie was really happy to be diagnosed with pulmonary tuberculosis. Before he arrived he was convinced he had lung cancer.
Robbie is in his late 50’s, he was born in Glasgow and left school at fifteen with no qualifications. His father was a shipbuilder on the Clyde. Robbie thought he would do the same, but in the seventies the shipyards closed* and there were no prospects for young men like him, so he came south.
He got various unskilled jobs, mainly in the china clay industry, got married and had two daughters. But he drank much too much and was not faithful to his wife, who left him. He says he does not entirely regret it – he had a great time.
Or as George Best famously said “I spent a lot of money on booze, birds and fast cars, the rest I just squandered”
Nationaal Archief Fotocollectie Anefo
Then, about three months ago, he developed a cough that kept him up at night. He had terrible sweats and would wake up in the morning with his bedclothes really wet. He took the sheets off his bed to hang them up to dry during the day. He knew something was seriously wrong when he started coughing up blood and was losing a lot of weight. He had always been “wiry” but now was becoming skeletal. Robbie became increasingly sure he had lung cancer, but was too frightened to get help. He still talked to his daughters every Sunday on the ‘phone. Last Sunday one of them was so worried when he told her what was happening that she went to find him and brought him in to our hospital. He had a chest Xray and within a couple of hours we told him that we thought he might have TB.
pulmonary TB usually affects the upper part of the lungs
We put him in a side-room and got him to cough up some blood-stained sputum which we sent to the lab. Within a couple more hours we got a phone call to say that the sample was “teeming” with AFBs – acid-fast bacilli.
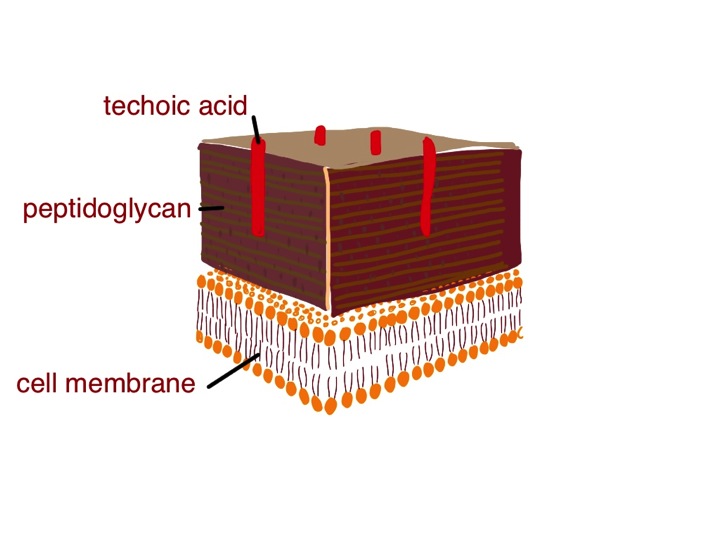
I have previously talked about bacterial cell walls and the Gram stain, which is a process which involves staining a biological sampe with a blue and red stain which have different affinities for the various components of a bacterial cell wall. Gram-positive bacteria stain bluish-purple with this stain because their cell wall is mainly peptidoglycan. Gram-negative have a thin layer of peptidoglycan, but a thick covering layer of lipopolysaccharide (LPS), which, like peptidoglycan, helps protect the germ from damage. As its name suggests, LPS is made of chains of sugar molecules and lipids – with carbon chains about 16-20 carbon atoms – similar to those in triglycerides in butter and olive oil. Gram-negative bacteria look red under the microscope after the Gram staining process
Tuberculosis bacilli stained with auramine and counterstained with acridine orange – viewed under uv light the mycobacteria shine out – Image from US Centers for Disease Control and Prevention –
Tuberculosis is caused by the bacterium mycobacterium tuberculosis. It has a quite different cell wall structure which makes it very difficult for our white cells to destroy. The secret weapon is mycolic acid. This a long molecule composed mainly of hydrocarbon chains – up to 54 carbon atoms long.
Hydrocarbon chains are common in all sorts of lengths in nature, most of which are useful to us. One carbon atom, surrounded by four hydrogen atoms is methane – a gas which I rely on to cook my dinner. Three or four carbons and we get the gases propane and butane, also useful in cooking the barbecue. Octane, a liquid with eight carbon atoms gets me to work in the morning. When we get to 16-20 carbon atoms there are endless things I can get from the supermarket, mainly to eat, with this size of hydrocarbon chain. These are mainly liquids or soft solids such as olive oil, butter, meat fat, and cooking oil. With longer hydrocarbon chains we are talking about waxes. Waxes have huge numbers of use in nature and are used extensively by humans, but mainly for protection rather than as a food. We use waxes a lot for protection – shoe and furniture polish for instance.
wax is used to protect shoes, furniture and a whole lot of other things
The wax in our ears is a mixture of squalene, a hydrocarbon of 26 carbon atoms mixed with cholesterol and dead skin cells, also designed to protect us from microbial infection. Plants produce waxes to protect the surface of their leaves, and sheep produce waxes to protect their wool (lanolin, chemically similar to earwax). Waxes are protective because they are difficult to break down chemically. The tuberculosis bacteria use this property of wax to protect them from the nasty chemicals white blood cells use to try to destroy them. We can detect mycobacteria by using a special stain which is taken up by the waxy coating. The original stain was called Ziehl–Neelsen, or ZN, but we now use a better one called auramine. This is a fluorescent dye which sticks to the wax, even when the specimen is flooded with acid and alcohol, which will remove it from just about any other bacteria which do not have mycolic acid in their cell wall. The specimen is then viewed under ultraviolet light and the TB germs light up.
Pulmonary TB is transmitted from one human to another mainly by coughing – producing a fine spray of droplets which contain the tuberculosis bacteria. Robbie mixed with a number of alcoholics and drug addicts, who are more prone to develop this disease. When the bacteria are detected by the cells lining the lung (see asthma and pineapple below), neutrophils will have a go at engulfing them and soaking them with lysozyme and bleach. TB germs will laugh at that (no, not really, TB germs don’t have much of a sense of humour). Their waxy coat makes them impervious to hypochlorite. Then the professionals are called in –macrophages. They will also engulf the tubercle bacilli. They will then call in help from T-lymphocytes, walling off the tubercle germs in a special structure called a granuloma.
cartoon of a tuberculous granuloma – the centre is caseating – cheese-like – made from dead macrophages
The TB germs can only grow very slowly, because they have to make the very long hydrocarbon chains to protect the new bacterium after cell division. Macrophages and lymphocytes use all the weapons at their disposal to try to kill them, many of which I think we don’t properly understand. We do know that nitric oxide, made by neutrophils is important. This is a gas which can penetrate the waxy coat and damage the TB germs. Another important molecule is thought to be granulysin made by cytotoxic T-cells. We don’t know how either of these molecules really do the damage and kill TB. Usually the human wins, but in about one in ten, as in Robbie’s case, TB wins. Robbie’s smoking and previous exposure to clay dust will have caused lung damage, which makes him more susceptible. His heavy drinking and poor diet will also make it worse. Some think vitamin D, which is made in the skin from cholesterol and sunlight might be important in defence against TB. Robbie likes dim, smoky rooms and avoids sunlight most of the time. Before effective drug therapy, sanitoriums would encourage patients to sit in balconies in the sun as part of their therapy, making lots of vitamin D.
Even when the human wins, it is not always a total victory. Sometimes there is an impasse, where the TB germs cannot be killed, but only constrained in the granuloma. In this siege scenario macrophages have another trick up their sleeve – calcification. Macrophages are very closely related to cells which make bone – osteoblasts. When the granuloma cannot be sterilised the macrophages start laying down insoluble calcium salts and imprison the TB germs. Often we see white spots on the chest xrays of elderly people who were exposed to TB when they were young – this is due to the TB calcium prisons which allowed their lungs to solve the problem of what to do with indestructible organisms.
It is quite likely that Robbie will recover from his TB, as long as he takes his drug therapy regularly, even though his lungs will end up even more damaged than they were. There are some very drug-resistant strains of TB emerging in India and Africa, but at present most TB in the UK is sensitive to rifampicin, isoniazid and ethambutol. He has promised his daughter that he will stop smoking and drinking so much, and eat better food.
This week’s food link is Edam cheese.
Edam cheese is case in wax to protect it from drying out and from spoilage. I think it is too bland, but my son likes it
This is traditionally encased in a wax coating to protect it from drying out and from microbial spoilage. I do not really understand why micro-organisms have not yet worked out how to easily break down wax and use its energy for their benefit. It has the same calorific value as fat but is clearly unappetising for all the normal germs in my kitchen which are happy to feed on shorter chain hydrocarbons.
*There is a very powerful speech by Jimmy Reid, a Glasgow shipyard worker who was elected Rector of Glasgow University.
Chris is a self-employed builder. He takes a tablet of omeprazole and some gaviscon every evening to prevent heartburn, but otherwise has been healthy most of his life, and is looking forward to retiring in a couple of years – he is now sixty three. But last week was awful. On Monday morning he went to work as usual – his company is converting a large house into apartments. When he arrived at work he felt suddenly unwell and vomited up his breakfast of toast and jam.
Then he started to get really bad cramping pains in his belly, and had to rush to the toilet to produce large amounts of diarrhoea. Luckily they had put a new bathroom into the conversion a few days before. He ‘phoned his wife, who came and brought him home. He continued to feel really unwell with completely liquid diarrhoea every two or three hours, including through the night. Sometimes the diarrhoea contained dark red blood. His wife wanted to call the doctor, but Chris hates making a fuss.
“It’s just a tummy bug” he told her, “I’ll be fine soon”.
After two days the doctor was called and found that Chris was really not very well. His pulse was fast and his blood pressure was a bit on the low side. He strongly suggested that it would be best for him to go to hospital for a drip, but Chris was not at all keen. He agreed to drink some oral rehydration made up from sachets the doctor had left him, and gave a blood sample. When the GP ‘phoned a few hours later, Chris’s wife told him that he had not been able to keep the salt solution down.
“I’m calling an ambulance – just persuade him that he has to go” the GP said.
When he arrived at the emergency department we had the results of the blood sample – his plasma creatinine was over twice the upper limit of normal – 260 micromoles/litre. His haemoglobin was a bit low- 105g/l and his CRP was very high at 180.
The ED department doctors had persuaded him that he had to be admitted and wrote “infective gastroenteritis and AKI” on the clerking form. AKI means acute kidney injury. I don’t like the blanket use of this recently fashionable term for any sudden increase in creatinine or drop in glomerular filtration rate. In this case Chris’s kidneys are not injured – they are doing their job very well, under difficult circumstances. By stopping urine output they are saving his life – if they had carried on producing the normal amount of urine despite severe dehydration Chris would most likely be dead. When an athlete runs 100metres and is very short of breath we don’t say he has acute lung injury – his respiratory system is responding appropriately to physiological demand – as were Chris’s kidneys.
He was put into a side-room on the acute medicine unit and his intravenous fluid was continued. His pulse, blood pressure and urine output were monitored. A couple of days later the nausea and abdominal pains had settled, his diarrhoea had reduced to twice a day, and he was starting to feel hungry again. The microbiology lab ‘phoned to tell us that they had found Campylobacter in the pot of blood-stained, watery liquid labelled as a stool sample we sent when he was admitted.
Where did the Campylobacter come from, and why did his wife not get it?
Campylobacter are helical, like H pylori – it helps them swim through the mucus layer and invade into the intestinal epithelium By De Wood, Pooley, USDA, ARS, EMU. [Public domain], via Wikimedia CommonsCampylobacter jejuni is a spiral-shaped bacterium and a relative of Helicobacter pylori which I have talked about previously (vomiting blood and sweetbreads). In the UK raw or undercooked chicken is the most likely source. It does not cause disease in the chickens, but is a commensal organism – amazingly about 60% of chickens reared in this country contain campylobacter in their intestines. Chris likes to cook, and a couple of days before he became ill had prepared one of his favourite meals – fried chicken and rice. He admits he might not have been as careful as he should about washing his hands and cleaning the chopping board after jointing the chicken – he will be in the future.
Most chickens bought in the UK contain campylobacter
Chris was particularly at risk because he was taking omeprazole, which reduces stomach acidity. The low pH in our stomach is important in killing nasty, pathogenic germs in food we eat and stopping them getting into our intestines. Nitrate secreted into saliva is also important – it is converted to nitrite on the surface of our tongue by bacteria which normally live there. This nitrite is swallowed and reacts with stomach acid to produce nitric oxide – which is toxic to many germs including campylobacter.
Chickens do not get ill from campylobacter – it is a commensal in their intestines
Stomach acid also has other uses. In many parts of the world people are lactose intolerant and get a lot of their calcium from eating bones rather than dairy products. I am told that when chicken is eaten in Africa and China, the smaller bones are crunched and swallowed. Stomach acid is necessary to dissolve bones and release the calcium.
The next question is why do some bacteria, if they get past the stomach’s defences, cause vomiting and diarrhoea, when our colon is full of bacteria all the time? Why do our resident colonic bacteria not stimulate an inflammatory response?
There are two main reasons – invasiveness and bacterial toxins. Like the stomach, the lining of the small and large bowel is covered with a layer of mucus, secreted by goblet cells. Our resident bacteria are well-behaved citizens and do not try to invade across the mucus layer into the gut epithelial cells. Mild incursions are allowed, but if they get as far as the base of the epithelial cells, they will trigger an alarm. These epithelial cells have toll-like receptors on the basal and lateral surfaces which, when triggered, will cause the police, in the form of neutrophils, to be called to the scene. The neutrophils will hunt down and digest these wrong-doers, causing a bit of havoc in the process. That is what inflammation is – the action of our immune system to destroy threatening pathogens. Campylobacter is not well-behaved, but attacks gut epithelial cells to produce inflammation. The result is leaking out of large amounts of fluid from the gut wall and stimulation of intestinal smooth muscle. Together these caused Chris’s abdominal cramps and diarrhoea. The inflammation can sometimes be so severe, as with Chris, to cause bleeding from the inflamed gut mucosa – bloody diarrhoea is known as dysentery. Some unfortunate patients with ulcerative colitis have inflammation and diarrhoea when there is no obvious pathogen – either something has gone wrong with the police force (immune system), or there is a pathogen which we have not yet found.
Many germs which cause gastroenteritis, including campylobacter, produce a bacterial toxin. I warn our students to be careful of using the word toxin, unless describing something nasty made by a bacterium. At least we should know the chemical name of the toxin. Alternative medicine therapists use the word toxin all the time, but I’m not usually sure what they are talking about, and you have probably guessed I’m not a fan.
The most dramatic effect of a toxin-producing organism is seen in cholera. This organism makes a toxin that interferes with the normal mechanism for absorbing water from the gut. Typically, patients with cholera produce more than ten litres a day of very watery diarrhoea, and if not given lots of oral or intravenous fluids will die from dehydration. There is an interesting story about sporadic cholera outbreaks happening in the 1970s, apparently at random in communities with good sanitary facilities. It was found that these occurred under the flight-path of aeroplanes flying from cholera-endemic areas. The planes were discharging the contents of their sewage waste tanks in mid-air at 30,000 feet to reduce weight. The cholera organisms were surviving the drop and causing mini-outbreaks of disease. The airplanes don’t do that any more.
Another toxin-producing bacterium we commonly see in hospitals is Clostridium difficile diarrhoea. It used to be very common in frail, elderly patients given broad-spectrum antibiotics. C difficile would frequently kill these patients in a thoroughly depressing, undignified manner. It is called C. difficile because, when originally identified as the cause of pseudomembranous colitis, it was very difficult to grow. This was because it is an obligate anaerobe. That means even very small amounts of oxygen inhibit its growth, but now we know this we can easily grow it in the lab. We can also identify the toxin, and detect C.difficile DNA using rapid PCR testing. We now see much less C.difficile diarrhoea – due to a combination of obsessional hand-washing and more careful use of antibiotics. It is worth knowing that the alcohol hand rubs do not kill C.difficile spores – when the lab are trying to isolate C.difficile, the first stage is to flood the specimen with absolute alcohol to kill off all the other bacteria. Washing with soap and water works by removing spores from the hands, not by killing them. Clostridium is closely related to anthrax, another nasty organism which makes almost indestructible spores. Clostridium botulinum also produces a very unpleasant toxin which can cause death by paralysis – interestingly nitrite is added to help preserve meat and prevent the growth of C.botulinum organisms.
salami contains nitrite which helps kill C. botulinum spores (and makes it pink)
This week’s food link is red kidney beans. They are fine if they are cooked in chilli-con-carne. But if they are eaten uncooked they can produce a nasty illness which resembles bacterial gastroenteritis – vomiting, diarrhoea and abdominal pain. It usually happens only 2-3 hours after eating raw or undercooked beans. Bacterial gastroenteritis typically causes symptoms between one and two days after eating infected food. Red kidney beans contain a toxin known as phytohaemaglutinin. This is destroyed by boiling the beans for at least 10 minutes. Undercooking (such as in a slow cooker which does not reach boiling point) can actually increase the amount of toxin.
Red kidney beans contain a toxin, phytohaemaglutinin which causes diarrhoea and vomiting if the beans are not cooked for long enough or at a high enough temperature.
Phytohaemaglutinin is one of a family of proteins, called lectins, produced by plants which selectively bind sugar molecules. The notorious spy-poison ricin, derived from castor oil seeds is a lectin. These proteins have many uses in medical research and in the past were used to identify the sugar molecules on red blood cells allowing us to type blood into A,B and O groups. Binding sugar molecules on the intestinal epithelial cells triggers increased secretion of fluid and smooth muscle contraction in a similar way to invasive bacterial or viral pathogens, but the illness following eating raw kidney beans normally only lasts a few hours, rather than days. But I would still encourage you to cook your beans well.
Diabetic ketoacidosis (DKA) is one of the more dramatic problems we deal with regularly on our acute medical unit. Ryan didn’t even know he had diabetes before he came in, although the possibility had gone through his mind. He is nineteen and is studying economics at university. Last weekend he came home from college. His mother immediately noticed that he had lost weight. She thought it was because he was not eating properly – but Ryan was insistent that he ate loads. He freely admitted his diet was mainly “rubbish” – lots of burgers and chips, and he drank loads of fizzy drinks (he does not like water).
Ryan would remove the green bits before eating this
Then on Saturday night he began to get abdominal pain and vomiting. Mum thought at first that the problem was that he had eaten some undercooked sausages and had too much to drink at a party at his friend’s house the day before.
On Sunday she and Ryan’s father became really worried. He was clearly more unwell, and his breath had a really odd smell – sweet and fruity. And he was staggering to the bathroom every couple of hours to pass urine or vomit again, or both. It was when he was too ill to get to the bathroom and peed in his bed that dad called the emergency out-of-hours doctor. The GP realised straight away what was going on. Ryan was breathing deeply and heavily, dried saliva and vomit was crusted round his mouth and he was very dehydrated and sleepy. She tested his blood for glucose – the reading was “hi” – off the scale. He was soon with us in hospital.
We have a protocol for dealing with DKA which involves lots of intravenous fluids – starting with normal saline, a continuous intravenous insulin infusion, and frequent and regular checks of blood acidity, glucose and potassium. We also look for any infection which might have precipitated the DKA.
There are lots of interesting and difficult questions to ask about type 1 diabetes, such as
– what causes the pancreatic islet cells which make insulin to be destroyed?
– why does this type of diabetes run in families?
But today I am going to concentrate on two easier questions:
– why do type 1 diabetics lose weight before their disease is treated with insulin?
– what causes ketoacidosis?
When I ask “why do diabetics lose weight?” the answer I often get is that they use up fat because their bodies cannot use glucose.
Glucose used to be thought of as a wonder food
This answer does not really satisfy. When we eat food our digestive system is very good at extracting energy. We are all familiar with the calorie content of food. One calorie (really kilocalorie or kcal) is the energy required to heat a litre of water by one degree centigrade. The energy content of food, measured in kilocalories or kcal, can be accurately measured by putting the burger and chips in a device known as a bomb calorimeter. This combusts the food with oxygen and measures the rise in temperature of the surrounding water bath. Let’s say the burger and chips, the three litres of cola, the cornflakes, milk and pork pie and other sundries that make up Ryan’s average daily food intake contains 2500kcal, if we were to put it in a bomb calorimeter.
bomb calorimeter – the bomb is the small silver cylinder- picture from wikimedia commons – by harbor1
Typically we absorb more than 95% of the available calories from an average meal. The only energy we cannot absorb is that in cellulose fibre*. Ryan does not eat much of that. Pretty much all of the 2500kcal he eats every day will be available as energy. Ryan is a fairly active young man. He uses 1500kcal a day just sitting studying or watching “man v food” on TV. Walking, running, cycling and energetic evenings with his new girlfriend consume the other 1000kcal. So why is he now losing weight? Weight loss means loss of fat. This is stored all around our bodies as adipose tissue. Adipose tissue contains cells that essentially only contain a large blob of triglyceride (we’ll talk about that later). Triglyceride contains nine kcal of energy per gramme, so if fat disappears, the energy must be going somewhere. The second law of thermodynamics says that energy cannot be created or destroyed.
The best way of thinking about it is that energy is going in – 2500kcal/day – and energy is going out – basal metabolic rate and energy used for exercise – also 2500kcal/day. If these are balanced, then fat stores will not change. Clearly, if Ryan is losing weight and using up fat stores there must be an imbalance with this input and output equation. The problem is not with the input, he is eating loads and absorbing the energy. It’s not with his basal metabolic rate, that is normal. It’s not with energy expenditure with exercise – that has not changed. The energy loss is in Ryan’s urine – it is full of glucose. Glucose has four kcal per gramme. He is peeing out about 800kcal per day of energy – that is why he has lost weight. It is also why he is passing so much urine and become dehydrated. Glycosuria causes an osmotic diuresis – causing him to lose extra water and be very thirsty and drink lots of fluid. Unfortunately although the fizzy drinks have lots of sugar, it is going in one end and out the other. Once the vomiting started, he was not able to keep up with the input and became even more dehydrated.
Now the ketoacidosis. This has got very little to do with glucose – it has everything to do with fat metabolism. Insulin is the hormone which controls energy metabolism. When we eat, carbohydrate is turned into glucose. Blood glucose is monitored by pancreatic islet cells. pic If glucose is high, the islet cells release insulin. Insulin has lots of important effects on how energy is used and stored.
When we are starved, with no carbohydrate intake, insulin levels drop and the lack of insulin:
1) promotes the conversion of glycogen (a glucose polymer stored in the liver) to glucose
2) keeps blood glucose levels acceptable by promoting the conversion of protein to glucose and
3) promotes breakdown of fat to provide energy.
The first two actions are important in keeping blood glucose high enough to maintain brain function– some brain cells absolutely rely on there being some glucose.
The third effect of low insulin –lipolysis – is also helpful in non-diabetics, breaking down fat to supply energy. Most tissues apart from the brain are very happy to get their energy from fat metabolism.
The problem comes when insulin is not just low, but completely absent. This never happens in non-diabetics. There is always enough insulin to keep fat breakdown under control. When insulin levels do go down to zero, in type 1 diabetics, fat starts to break down very quickly.
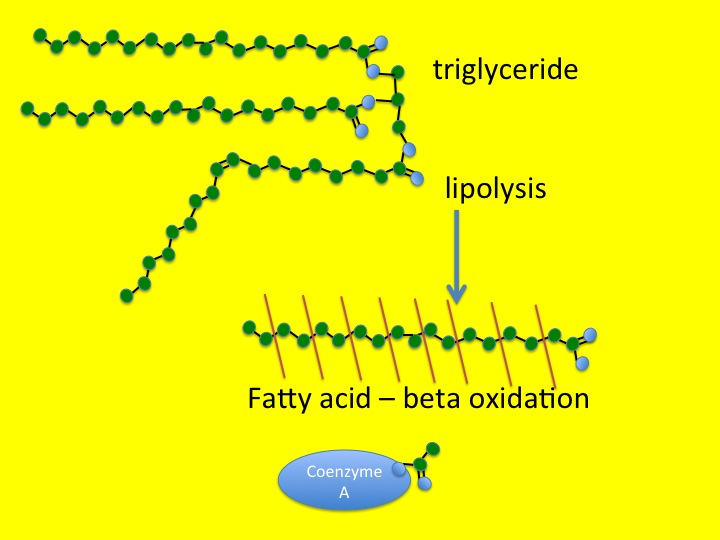
This process starts as lipolysis – the removal of the long fatty acid side chains from triglyceride. Triglyceride is an ester of fatty acid and glycerol. The glycerol backbone can be used to make more glucose (not that it is at all needed). Then the long-chain fatty acids are broken down, two carbons at a time. This is known as beta-oxidation.
triglyceride is first broken down by lipolysis, which removes the fatty acid chain – the fatty acid is then chopped up two carbons at a time by beta oxidation, attached to coenzyme A is then processed into carbon dioxide, water and energy by mitochondria
You will remember from previous posts (chest pain and horsemeat lasagne) that the carbon atom at the other end from the carboxylic acid group in a fatty acid is known as the omega carbon – hence omega 3,6 and 9 fatty acids which have double bonds in those positions. The carbon at the acid end of the molecule is the alpha carbon, and the next one along is, of course, a beta carbon. Hence beta-oxidation oxidises this carbon and removes an acetyl group from the fatty acid and attaches it to a Coenzyme A molecule. This can then be fed into the Kreb’s cycle and electron transport chain to convert it into carbon dioxide, water and nine kcal/gramme of energy in the form of ATP.
When insulin levels are very low, as was the case with Ryan, the rate of lipolysis and beta-oxidation are so fast that the Kreb’s cycle cannot keep up.
lipolysis is controlled by insulin levels in the blood – in non-diabetics insulin secretion does not drop below about 0.5 units/hr – type 1 diabetics can have no insulin and lipolyisis is unrestrained
Fuel is being delivered at a faster rate than it can be burned. The irony of the situation is that there is plenty of fuel around in the form of glucose, but because it is not triggering an increase of insulin, the fat cells do not know this and keep on breaking down triglyceride. The only thing to do with all this broken down lipid is to make ketoacids (also known as ketones). In small amounts these are a quite useful alternative to glucose for energy production when we are starving – there are measurable amounts in blood and urine in healthy people if we do not eat for 12 hours or more, but low-level insulin secretion keeps the amount in check.
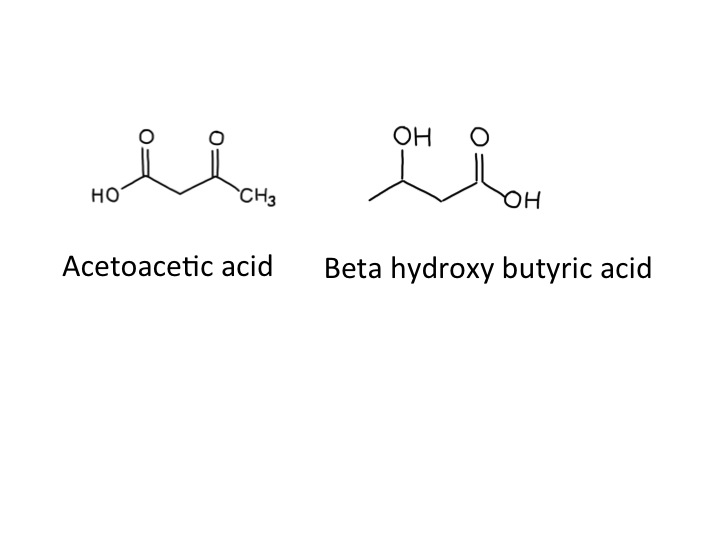
When ketoacids are made in huge amounts they cause problems because they are acidic. The two ketoacids we make are acetoacetic acid and beta-hydroxybutyric acid. Acetoacetic acid slowly, spontaneously degrades to make acetone and carbon dioxide,
acetone is commonly used as a solvent
which was why Ryan’s breath smelt sweet and fruity – like nail polish remover. I think that beta-hydroxy butyrate is not technically a ketone – but it is an acid. I don’t think it is necessary to make a fuss about this (but the pedant in me forced me to mention this). Acidosis makes people ill – our bodies are designed to work with a blood pH of between 7.35 and 7.45. Ryans blood pH was 7.04 when we first tested it. Many of the enzymes in our body just don’t work properly when the blood is too acid – when it becomes too severe acidosis can cause drowsiness and then coma and then death. The other problem is that the physical stress caused by ketoacidosis and dehydration from high glucose result in the release of glucocorticoids (cortisol) and adrenaline from our adrenal glands, both of which further encourage lipolysis and the formation of more ketoacids. A potentially lethal viscious cycle.
How does neutral fat make acidic substances? Well, acidity is not like water, sodium or energy where what goes in must come out. We can drink lots of acidic vinegar and it will not cause any change in blood acidity. The easiest way to look at it is that when we make small, charged molecules from large uncharged fat molecules, the hydrogen atoms are happy to give an electron to the anion (such as acetate) and drift off as a positive hydrogen ion – ie increase acidity.
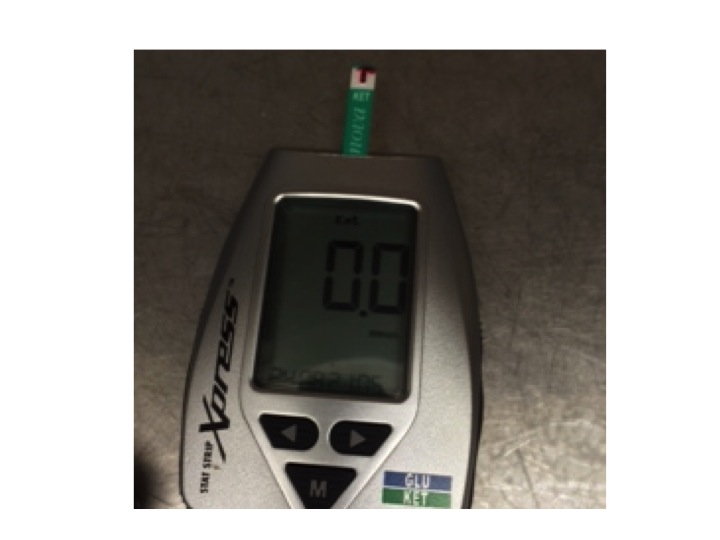
We always used to rely on measuring urine ketones, but now have a blood ketone meter which is much better at quantifying the amounts in the blood of patients with ketoacidosis. Ryan had a level of 9.6mM when he came in. By the next day he was sitting up, eating lunch, with a level of 0.3mM.
blood ketone meter – I measured my blood just after breakfast – that was why the reading was zero
We used to use a sliding scale to treat patients with ketoacidosis. This meant that the higher the blood glucose, the more insulin we gave. It is quite clear from what I have said about what causes ketoacidosis that this was not a sensible strategy. We now give enough insulin (>6units/hr) to completely supress lipolysis until the level of ketones in the blood becomes normal, and usually have to give extra glucose to prevent blood sugar levels going too low.
Now to the food link – butter. As previously discussed, butter is mainly made from triglyceride. It is yellow because it contains vitamin A, or carotene. This helps prevent it becoming oxidised, or rancid. When butter does goes rancid, it is because the fatty acid chains become oxidised by bacteria, in a process similar to beta-oxidation. One of the main oxidation products of butter is called butyric acid. And, amazingly, the reason butyric acid is called that is because it was first identified in rancid butter – Latin for butter is butyrum (cow-cheese). The simple hydrocarbon with four carbon atoms then became known as butane, again named from butter/butyrum. Not sure why methane, ethane or propane are called that. If I find out I will let you know in a future post.
*cellulose is a glucose polymer made by plants to provide structural support – when it is eaten as food it is known as fibre – it burns well in a calorimeter but cannot be used by humans to provide useful calories – but can be used as an energy source by cows, which, of course, provide us with butter. Ryan likes butter but he does not like fibre. The diabetes dieticians are trying to change that. I wonder if he will be eating brown-bread toast now? (see my very first post – toast and diabetes).
We admit a lot of elderly patients who have fallen over. There are a whole range of problems which cause falls, one of which is postural, or orthostatic hypotension. Emily is 78 and lives alone in a bungalow. Her husband died three years ago, but her daughter calls in to help most days. She was fairly well until she went to her family doctor’s surgery last week and had her blood pressure measured. The result was not good – 186/88mmHg.
She first developed high blood pressure when she was in her fifties, and it had crept up since. She was already taking ramipril (an ACE inhibitor) and indapamide (a thiazide diuretic). She had tried amlodipine, but that gave her swollen ankles, which she hated. Her GP decided to try her on doxazosin. This is an alpha-blocker that lowers blood pressure by opening up small arteries. The morning of the day of admission she got up and took her tablets. Then she had a bath, but when she got out felt awful, and faint, and fell to the floor. She lay on the floor for a few minutes and tried to get up, but felt very dizzy and had to lie down again. She phoned her next-door neighbour on her mobile phone. The neighbour came round and helped Emily to her feet, but she went wobbly and could not stand, ending up on the floor again. Her neighbour became really worried and called for the ambulance. When the ambulance paramedics arrived they found that when they tried to stand Emily up, her blood pressure dropped from 160mmHg systolic to 90mmHg. They brought her in to see us.
There is a popular notion that all antihypertensive drugs cause postural hypotension, but this is not the case. As we will see, beta-blockers can even be used to treat this condition.
What is happening with the circulation to cause Emily’s blood pressure to drop when she stands up?
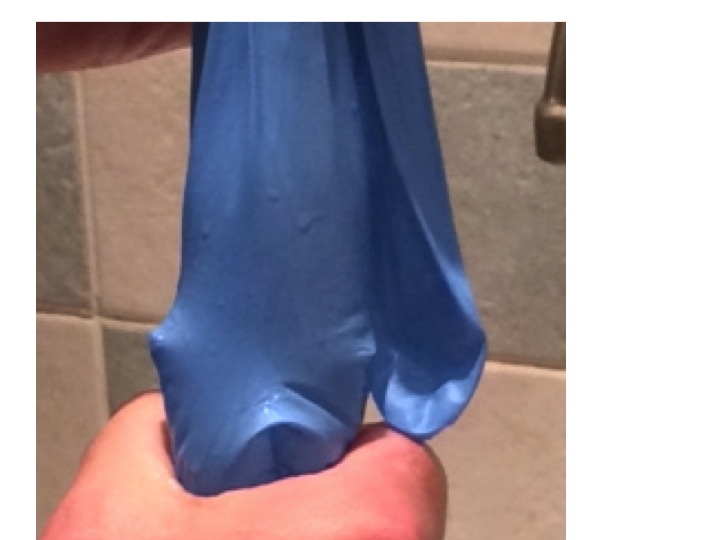
standard clinical rubber glove
The whole explanation is made much easier with a standard hospital rubber glove (the stretchier the better) and a sink, and a supply of water. First we fill the glove up with the right amount of water.
glove about half filled with water – laid flat – notice the thumb is filled with water – this represents cardiac filling pressure
The water in the glove represents blood in the venous side of the circulation. The arterial side is much lower volume, and is very little affected by changes in pressure due to posture.
When we stand up the amount of fluid in the veins of the legs and abdomen increases, due to increase in hydrostatic pressure stretching the rubbery material. Veins are thin-walled and rubbery, and will naturally distend if the pressure inside is increased. If this happens, the thumb empties, this represents the filling pressure to the heart. If cardiac filling pressure drops, cardiac output drops and blood pressure falls.
no venous squeeze due to failure of the autonomic system, due to brain stem or peripheral sympathetic nerve action means no cardiac filling pressure – the thumb is empty
This will result in a fall in systemic arterial blood pressure – postural hypotension.
But this does not happen in healthy, young people. We have autonomic reflexes which cause constriction of veins before we stand up, so that filling pressure to the heart is maintained. In the glove model this is represented by my hand squeezing the fingers and main hand of the glove.
in healthy individuals the autonomic nervous system provides a squeeze of abdominal and leg veins to maintain filling pressure of the heart – notice the thumb is now full of water
There are two main causes for postural hypotension – failure of autonomic squeeze of the veins, and insufficient filling of the venous compartment.
Let’s take the first of these – not enough squeeze. When we move from lying down to standing up, our brain knows what is about to happen. As we move our limbs the proprioceptors in our joints tell our brain what is about to happen and our conscious mind probably also has a role. Messages are sent to the control centre (Houston – prepare for lift-off) in the medulla. Sympathetic nerves send messages down to the veins to tell them to constrict. It is interesting that human intra-abdominal mesenteric veins are particularly richly supplied with sympathetic nerve endings – presumably because we are one of the few animals where standing is such a problem. In animals such as horses and dogs, moving from lying to standing does not involve a capacious venous network to be subjected to large pressure changes – their legs have very little venous blood and their abdomen and contents is on the same level as the heart.
It is also not surprising that if you take a healthy, young person and strap them to a tilt-table, and suddenly move them from horizontal to vertical, they will experience an impressive drop in blood pressure. If they are lying on a bed and stand up – blood pressure hardly changes because the brain-stem prepares us for the change in posture with messages to our abdominal and leg veins to constrict.
As we get older, everything starts to go wrong (see last week’s post). Autonomic reflexes become impaired. Patients with Parkinson’s disease are particularly prone to develop postural hypotension because of impaired brain stem autonomic reflexes – this used to be called Shy-Drager syndrome when I was young – it is now know as multi-system atrophy. Some drugs such as antidepressants and methydopa inhibit brain stem sympathetic output and predispose to this condition.
The sympathetic nerves end up on the outside (adventitia) of veins and release noradrenaline which causes constriction of venous smooth muscle by stimulating alpha receptors. Emily had been given an alpha-blocker to reduce constriction of her arterioles and thereby reduce blood pressure. In her it reduced the ability of veins to constrict when standing up by blocking venous alpha receptors.
Some patients have problems with the autonomic nerves. Diabetics and alcoholics are prone to develop autonomic neuropathy which may result in postural hypotension due to inability of veins to constrict.
Temperature also plays a part in venous constriction. Most of us have noticed occasionally when getting out of a hot bath or sauna feeling dizzy for a few seconds, we hold onto the edge of the bath then things improve. What has happened is that the hot bath water has caused venous dilatation and the normal reflexes have not been enough to maintain cardiac filling pressure. Cardiac output drops, baroreceptors panic (Houston, we have a problem). Houston responds quickly by sending a stronger sympathetic signal to the veins and to the heart to increase cardiac output and the problem is soon sorted. But of course preventing the problem is much better than reacting when it has happened.
The second main cause of postural hypotension is insufficient fluid in the circulation.
to represent fluid loss – some water is poured out of the glove
In Emily’s case this may have been partly due to the thiazide diuretic she was taking for her blood pressure. Patients on loop diuretics such as furosemide are even more prone to fluid depletion. Sepsis causes problems with a postural blood pressure drop because of fluid shifts out of the circulation into interstitial spaces, and because fever causes venous dilatation as in the hot bath above. Jonathan, who had a lobar pneumonia a few weeks ago, may well have collapsed in his GP’s surgery because of a postural blood pressure problem.
even with reduced volume – when lying flat the thumb is still filled with water and blood pressure is maintained
Blood loss from trauma or intestinal bleeding may often not result in blood pressure drop until the patient stands up – it is far easier to maintain filling pressure when lying down and not having to squeeze the veins hard.
with reduced volume, squeezing the legs and abdomen no longer maintains filling of the thumb – cardiac filling reduces and blood pressure drops
Sorting out Emily’s postural hypotension was straightforward. We gave her two litres of intravenous saline and stopped her doxazosin, and diuretics. We had a plan to start her on verapamil or diltiazem when she could stand up without her blood pressure dropping – which it did the next day.
Do ACE inhibitors/ARBs, beta blockers or calcium channel blockers cause postural hypotension?
The answer is to begin with, no and no.
ACE inhibitors can cause a postural drop when they are first started. This is because angiotensin does have an effect on venous constriction, but only transiently when it is increased or decreased. If angiotensin II is infused into a hand vein it will constrict, but only for a few hours at most. Similarly if angiotensin II is withdrawn, the vein will dilate, but only briefly. This means that it is prudent to warn patients that they may develop postural symptoms following the first dose – and it is a good idea to take it when lying down before going to bed. The effect of angiotensin II on arterioles is long-lasting, which is why these drugs are useful in treating arterial hypertension.
Beta blockers reduce blood pressure by mechanisms which are not completely understood, but is likely to be a combination of reduced cardiac output and inhibition of renin release from the kidney. There are beta2 receptors in arteries and veins, but these cause vascular dilatation. Beta blockers can help veins constrict, which is why they are sometimes used to treat postural hypotension. Having said that, if there is a postural drop in blood pressure, the reflex mechanisms set in train by baroreceptors involve increased sympathetic stimulation to the heart via beta1 receptors, increasing force and rate of contraction. Beta blockers will block this response and may therefore impair the recovery from an episode of postural hypotension, but they will not cause it.
Calcium channel blockers have an effect on arterioles, not on veins. Calcium channels are important in maintaining arterial tone, but not venous tone. Venous tone depends almost entirely on sympathetic stimulation. Therefore, at least in theory, drugs such as verapamil, diltiazem and amlodipine will not cause a postural drop. But of course if there is a postural drop, lower initial blood pressure caused by these agents may make the episode worse.
Sorting out Emily’s orthostatic hypotension was fairly straightforward. What about when it is caused by age-related impairment of brain stem function, or by irreversible peripheral autonomic neuropathy? It can be a real problem.
overfilling the glove means that even with no squeeze to the veins the thumb remains full of water when the glove is upright – overfilling the circulation with fludrocortisone can help prevent postural hypotension
One approach is to use fludrocortisone. This is a synthetic analogue of aldosterone. Made from cholesterol (again!) this hormone is made in the adrenal cortex, in different cells which make cortisol from cholesterol. The function of aldosterone is to regulate how much salt and water is excreted from our kidneys. Aldosterone helps the reabsorption of more salt from the distal convoluted tubule and will therefore increase circulating volume of blood. This will have the opposite effect to diuretics and overfill the circulation and help keep filling pressure at heart level adequate on standing up. Another drug which is sometimes used is midodrine, an alpha receptor agonist, helping constrict veins and maintain cardiac filling when upright. The drug does not have a licence for this indication in the UK.
We have made a not very good video showing how postural hypotension works – my first attempt at youtube – thanks for your help Steph. We will try to make a better one soon and update this post. The video is at:
The food link this week was not obvious when I started, but of course it has to be liquorice.
liquorice has been popular for a long time – it is made from the root of the liquorice plant
Liquorice contains glycyrrhizin which has a chemical structure similar to steroids such as cholesterol and aldosterone (made from cholesterol – again!). It has long been known to have a mineralocorticoid effect. This is not because it acts directly in the kidney to stimulate aldosterone receptors. Instead it prevents the conversion of cortisol to cortisone. Cortisone, synthesized by the adrenal glands, is converted to cortisol in the kidney, then back to cortisone, cortisol has more mineralocorticosteroid effects than cortisone – full details are in the NEJM paper:
Dorothy was born in 1911. She is 102. When someone over 100 is admitted to our ward there is always a lot of interest – but it is happening more and more. Dorothy, or Dot, as she is known by her friends, has been living on her own in sheltered housing. She has her own ground floor flat, but eats meals with the other elderly residents and has help with washing and dressing in the morning, and there is always a manager on hand if there are problems. She can make herself simple meals and cups of tea, and her 78 year-old son, David, usually visits every Sunday. I saw her in our ward last week. She had fallen over on Sunday night and bashed her head on the edge of a table, resulting in a huge bruise over her right eye. She has atrial fibrillation and is on warfarin – the ambulance crew brought her in to hospital as she was a bit confused and they were worried she might have a blood clot on her brain.
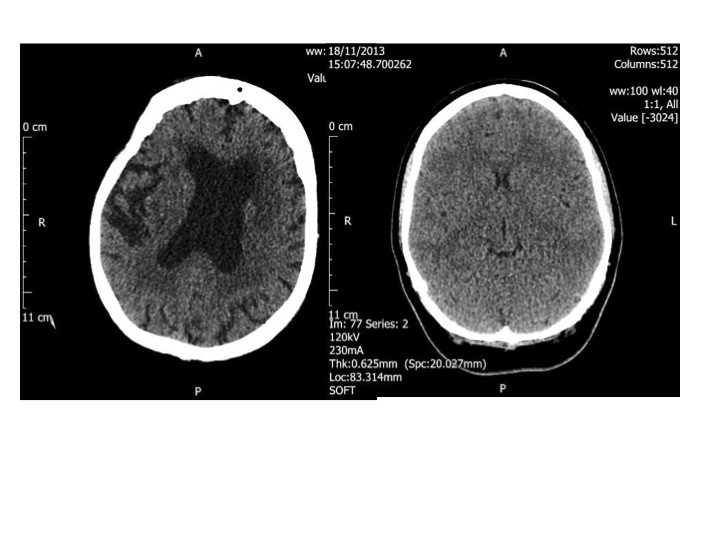
The CT head scan on the left is that of a healthy 96 year old woman, the one on the right is that of a 23 year old woman. The dark portion in the middle of the old brain is fluid, not brain. The younger person’s head is completely full of brain.
Luckily the CT scan of her head showed no evidence of serious bleeding, although her brain was considerably shrunken compared to a young person. She was quite deaf, but clearly had most of her marbles. She recounted how her father had fought, and was wounded, in the Battle of the Somme in the first world war, and her husband Rodney, who died in 1980 had fought in the second. She worked in a munitions factory during the war and had three children – all boys. It’s strange calling elderly people children, but offspring or any other word does not seem quite right. Only the youngest was still alive. She could remember the names of all 5 grandchildren and 7 great-grandchildren, but did struggle with the name of the Prime Minister and what year it was. Her brain was old as well as her skin and her ears, and all the rest of her.
It has been shown that for any cohort of elderly people, the main determinant of longevity is how old you are – as we get older the more likely we are to die sooner. The second most important factor is gender – women live longer than men. The third, somewhat surprisingly, is how fast you can walk. This is probably because being able to walk fast requires a lot of things to be working well – good lungs, good cardiac function, good muscles, good joints, good brain function, good eyesight, good peripheral nerve function etc. The paper suggests another alternative explanation – that if you walk slowly the grim reaper will catch up with you.
One explanation of why elderly people die soon if they cannot walk fast. Illustration by Belle Mellor – http://bellemellor.com (with permission – thanks Belle)
Why do people get old? This is a difficult question, and I cannot give you a definite answer, but will give you my thoughts. Ageing (aging in the US) seems inevitable. We can guess how old someone is by their skin, which becomes wrinkled – their hair becomes thin and grey, and their posture becomes bent. The cells in our skin, bones, brain and just about everywhere else continually become older, but some of our cells do not age in the same way. Think about a sixty year old man who becomes a new father. His sperm have contributed to make a baby that is not old, but brand new. The cells in his testes which have made the sperm are old, but they must have in them the information about how to make a cell which is not old. This, to me, very much suggests that old cells have not lost the information in the form of DNA sequence that they had when they were new. The creation of Dolly the sheep confirms this. Dolly was made from the nucleus of her “mother’s” mammary gland cell (she was called Dolly after Dolly Parton, who has very impressive mammary glands). In a test-tube this nucleus was inserted into an egg cell of another sheep (also her mother?) and processed to de-differentiate it.
Dolly Parton – from Wikimedia commons
This means the nucleus of the mammary gland cell was changed so that instead of only being able to make more, old, mammary gland cells, it could make all the new and different cells of baby Dolly the sheep. So, I’ll ask the question again, why do cells get old? It’s not that they have lost the information about how to be new cells, but nature has determined that after a certain period of time they change into old cells. What is happening in the cells to make them old? – clearly they are not so damaged that the DNA sequence is irreparably altered. There must be a clock inside cells that tells them how old they are – this is suggested by the classic studies of Leonard Hayflick, who showed that cells from human babies only divide about 50 times, and then stop.
cells must have some sort of clock inside them – it may well be the amount of methylation of DNA in the nucleus
Cells from older people divide far fewer times. It was thought for many years that the reason cells could only divide a certain number of times was because each time division took place, the teleomeres at the end of each chromosome become shorter. When they become too short, the cell can no longer divide. Teleomeres have been likened to like the plastic bits at the end of shoelaces – they stop the lace from fraying and are necessary in chromosomes to keep the DNA strands from coming apart.
It may well be that short teleomeres contribute to the cellular clock, but it seems they are not the only answer.
teleomeres are on the end of chromosomes like the bits of plastic on the end of shoelaces – to stop them fraying
More recent research suggests that epigenetic changes in DNA may be responsible. Epigenetics is a very fashionable science. Once a cell is differentiated in the growing embryo – say into a skin cell – its DNA becomes progressively altered. In particular methyl groups (CH3) become attached to a cytosine nucleotide when a cytosine and guanine are next to each other in the nucleic acid chain (CpG). This methyl group attached to the cytosine is copied when the cell divides. Recent research suggests that the age of cells is closely related to the number of methyl groups attached to CpG sequences in DNA.
What to me is really interesting, is that when sperm cells meet egg cells and their nuclei fuse to make the start of a new baby, all the methyl groups are stripped from the CpG sequences in DNA. This looks like a definite candidate for the cellular clock.
But why do we need to age? What if cells stayed “new”? What if we could stop the cellular clock? If we could inject a substance into Dot which reversed the epigenetic changes would she suddenly get lovely wrinkle-free baby-like skin and a fully functioning brain, ears and eyes? I don’t know, but I suspect not – answers please.
Why did evolution invent a clock in our cells to stop them dividing as we get older? One explanation for this is that cells become damaged with time, and dividing may not be a good thing for damaged cells as it is more likely that they will become malignant. We now know that most malignant cells develop a sequence of gene defects over time, which when added together cause the cell to become cancerous. Maybe the clock, which limits the number of divisions, is a way of preventing further DNA damage and malignancy. Certainly it seems that reactive oxygen compounds that can cause damage to DNA also cause CpG methylation – in a way causing premature ageing of cells.
I used to think that the reason our cells got older was so that we would die and no longer be competition for our children, who, through evolution, had become more fit than us. It seems this is not the explanation because there is very good evidence that early humans did not die from age-related causes, but from infections and trauma. The average life-span in prehistoric times was much lower than in modern times, most people dying before 30 and nearly all before 40 years of age. This means that it is not possible for evolution to select for, or against, those people who lived longer, because there were so few of them. Instead it seems that evolution only cares about keeping us alive until we have produced and reared babies – after that it has no interest or influence one way or the other. If there was a new gene that made a prehistoric human live longer, it would be extremely unlikely that the owner would survive longer and there would be no positive or negative pressure to keep it. This does not mean that our genes cannot influence how long we live. An important factor in determining how long we live is how long our parents lived. Another is where we live and our lifestyle.
Roger Corder, scientist and author of The Red Wine Diet
There is a wonderful book by an erstwhile colleague of mine, Roger Corder, called The Red Wine Diet, which suggests that eating foods with certain polyphenols can make us live longer.
polyphenols such as tannins are large molecules made up of lots of phenols – they are useful antioxidants which trap oxygen radicals
Polyphenols are found in plants and are often brightly coloured. Tannins are polyphenols, and cause a characteristic “furry” taste in the mouth because they react with the protein molecules on the surface of our tongue. These molecules are powerful antioxidants. What does that mean, and why do plants make them? In the case of a grape, one of its main concerns is DNA damage by ultraviolet light. Although short wavelength UV light can directly damage DNA, it seems that less powerful, longer wavelength rays can generate very reactive oxygen radicals such as singlet oxygen and hydroxyl, which then combine with, and damage DNA. Polyphenols can rapidly “mop up” or detoxify these oxygen radicals and protect DNA. If you live in the mountains of SW France, Sardinia, Crete or Sicily, the plants, and especially grapes, are subjected to large amounts of UV irradiation and produce lots of polyphenols. The people who live eat and drink lots of these polyphenols live to an unexpected old age. It might be a coincidence, but, for me, the inconvenience, expense and sheer tedium of drinking three glasses of red wine a day seems worth a try. But don’t overdo it and do remember Kevin (vodka and sweetbreads) below. The food link this week is a drink – Malbec.
Argentinian Malbec is full of tannins
This is a grape variety, which, according to Roger Corder, contains very high levels of procyanidins – tannins which he thinks helps protect us from heart disease and cancer, and makes us live longer. Antioxidants prevent the oxidation of LDL cholesterol (see previous post on chest pain and horsemeat lasagne) and reduce DNA damage that can make normal cells malignant and age more quickly. It is grown in the mountains of SW France and is the main variety in Cahors wine – a particularly dark and astringent glassful. Argentinian Malbec is more available in the UK and is also grown high up in the mountains where it gets plenty of ultraviolet light.
This is the third in a series of rigors. Some doctors call them chills, but I think of a rigor as a really uncontrollable shaking episode. I remember when I was a junior surgical house officer I was looking after patients who had undergone prostate surgery. From the nursing station at one end of the ward, I would hear a bed shaking at the other end. The beds in those days had metal frames and really clanked loudly when someone had a rigor – which on the urology ward was due to gram-negative septicaemia.
“Could you please draw up some gentamicin and ampicillin?” I would ask the nurse as I walked down the ward, armed with blood culture bottles to see an elderly man looking very alarmed, shaky and unwell. I think these days we are more careful to make sure our patients do not have infected urine before operating on them.
Earlier this week we admitted Jonathan. A very fit 36 year-old man who had never been in hospital before, apart from when he had an arthroscopy for a damaged knee following a football injury. It started with him feeling off-colour, off his food, and generally aching all over. He thought he had the ‘flu. He woke up at four in the morning to find it really hurt on the left side of his chest when he tried to breathe deeply. He had never experienced pain like it before, even after the arthroscopy. Then he started shaking uncontrollably. He then began coughing, and that was even more painful. Then he coughed up some brownish-reddish, rusty-coloured dark phlegm. He got up and took some paracetamol, went back to bed, but could not sleep. He decided he could not go to work and went instead to his GPs surgery. He had another rigor while in the waiting room, stood up and collapsed to the floor. The practice nurse found a couch so he could lie down, and got the GP to see him straight away. He was with us a couple of hours later. By the time we got to see him he had had a chest X-ray which showed a left lower lobar pneumonia.
Pyelonephritis, ascending cholangitis and lobar pneumonia are the diseases which commonly cause severe rigors. Malaria is another we see less commonly in our part of the UK, but can certainly also produce impressive rigors. To get rigors you need to have a flood of microbes released over a short period of time, which happens if you have an organ such as the kidney, liver or lung full of germs. Other infections cause fever, but less commonly rigors.
The reason for shaking with rigors is that our body is trying to increase our temperature quickly. The mechanism is similar to shivering when we are cold – using muscle contraction to generate heat. Substances on the bacterial cell wall are recognised by Toll-like receptors on macrophages. This is part of the innate immune system which is designed to respond to a whole variety of germs. Toll-like receptors are on a range of white blood cells, intestinal epithelial cells, respiratory epithelial cells and vascular endothelium – all the places we need to keep a close eye on where the enemy is likely to invade. Lipopolysaccharide, the coating of gram-negative germs is the classic trigger. Techoic acid on the surface of gram positive organisms (see last week’s post) is another. These receptors can even recognise DNA sequences only found in bacterial but not in mammalian cells. When macrophages are alerted to the presence of bacteria, they generate cytokines such as interleukin 1 which circulate and are sensed by the hypothalamus in the brain. The hypothalamus is the part of the brain which regulates body temperature and coordinates the shaking response. When Jonathan started shaking he felt really cold, even though his body temperature was higher than normal. When we saw him in the emergency department he was wrapped in three blankets, even though it is always warm in there.
Why do we develop a fever with infections? There is some evidence that some bacteria such as Salmonella typhi, which causes typhoid fever, are less able to survive at 40 degrees than 37 degrees. Not sure it helps much with other germs.
The junior doctor took the history and examined Jonathan. He took blood cultures and prescribed intravenous co-amoxyclav and clarithromycin.
Making a diagnosis of lobar pneumonia is usually easy in hospital, when we have an X-ray to look at, although in the early stages the changes can be quite difficult to see. We are all taught as medical students what clinical signs to look for. I thought it would be useful to think about why they happen in patients with lung problems, and particularly pneumonia.
First it is worth thinking what the lungs are for. They do two main things: 1) get oxygen into our bloodstream from the air and 2) remove carbon dioxide from our blood so we can breathe it out. What happens when these vital functions don’t work properly? Normally our lungs are very good at getting oxygen into our bloodstream. Blood coming out of our lungs into the pulmonary vein and then into our main systemic arteries is usually saturated to 98%. We can measure this easily with a pulse oximeter. This is a wonderful device which has revolutionised our management of patients with respiratory conditions. It was invented in 1972 by Japanese engineers called Takuo Aoyagi and Michio Kishi. The way it works is that it shines two beams of light through the finger tip or earlobe. One beam is red light at wavelength about 610nm, the other is in the near infra-red at 910nm.
oxy and deoxy haemoglobin have different absorption characteristics, which makes them different colours – the sats meter measures absorption at two different wavelengths and compares them, but only in the pulsatile part of the signal
If you look at a pulse oximeter you can only see one red light, the infra-red is not visible.
Measurement of oxygen saturation relies on the absorbance of oxyhaemoglobin being different from deoxyhaemoglobin. We are talking about the colours of haem and pyrroles – again! Oxygenated arterial blood is well known to be redder than venous desaturated blood. The diagram shows that the absorbance spectrum of haemoglobin is very different at 610nm in the two types of haemoglobin. But at 900nm it is very similar. The machine compares the absorbance at the two wavelengths and can calculate the percent oxyhaemoglobin to deoxyhaemoglobin.
photo of pulse oximeter probe – interestingly it looks very differenet in the photo from real life because my mobile phone camera captures infra-red images which we cannot see with our eyes normally – try it yourself
If it just did that, we would find that the haemoglobin in a fingertip is normally about 70% saturated, because most of the blood is in veins, rather than arteries. The really clever thing about a pulse oximeter is that it analyses the ratio of oxy- to deoxyhaemoglobin just in the pulsatile part of the signal. As the pulsatile part is due to arterial filling of the fingertip, this gives arterial, rather than venous oxygen saturation. Jonathan’s arterial saturation was 91% before he was given oxygen therapy, when it rose to 98%. There is a common misconception that hypoxia will make you breathless. Chemoreceptors in the carotid body and brain stem can certainly respond to hypoxia, but only when it is very reduced. Flying at 30,000 feet in a jet plane is equivalent to being at 8,000feet altitude where the oxygen tension is reduced by about 20%. Passengers in jet planes will often have oxygen saturations around 92% but do not feel breathless. In fact hypoxia does not cause breathlessness unless it is really marked. Why is this? – The reason is probably because increasing the rate of breathing does not help. Without oxygen, if I asked Jonathan to breathe faster, firstly he would find it difficult because it was so painful, but even if he did, his oxygen saturation would not improve. This is because there is shunting of blood in the part of his lung affected by the pneumonia. In the inflamed part of the lung there is increased blood flow, but the air spaces are full of pus. There is consolidation. Any blood which flows through this part of the lung will not be oxygenated and will emerge into the pulmonary vein desaturated. Even if the rest of the lung is working perfectly, saturating the blood to 100%, there is no way to resaturate the desaturated blood which has travelled through the abnormal lung, so his sats meter will detect this and show a low reading. It looks like evolution has sussed this out and doesn’t bother to make us breathe harder – unless the oxygen saturation drops to the mid 80s when various tissues in our body start to complain and generate lactic acid.
Carbon dioxide is a different story. Every day we breathe out about one kilo or two pounds of carbon dioxide. Our lungs are not designed to get rid of CO2 completely. Jonathan’s blood gas analysis showed a normal pCO2 of 5KPa (normal range 3.5-6). We need a certain amount of carbon dioxide in our blood to keep the acidity normal. But most (80%) of the carbon dioxide is in the form of bicarbonate (HCO3), made from carbon dioxide by the enzyme carbonic anhydrase in red blood cells.
carbon dioxide is in equilibrium with acid and bicarbonate
If our lungs are not working, then carbon dioxide levels in the blood increase, bicarbonate increases, and hydrogen ion concentration increases, because of the balance between carbon dioxide and bicarbonate (see equation). We are all familiar with the reaction of bicarbonate and acid (vinegar or lemon juice) to produce carbon dioxide.
bicarbonate and vinegar
This is a reversible equilibrium reacation and in blood this can work the other way round so carbon dioxide makes acid and bicarbonate. This is known as respiratory acidosis. Unlike oxygen, our body is very sensitive to carbon dioxide levels. This is first detected in the carotid body, located at the division of the common carotid artery into internal and external branches, either side of the thyroid cartilage. This acts as an “early warning” signal, sending messages to the brain stem that
when mixed together produce lots of carbon dioxide
something is amiss and the lungs aren’t working properly. A few seconds later blood reaches the medulla in the brain stem, which is the central control mechanism for breathing, next to the vasomotor centre controlling the circulation. High carbon dioxide also stimulates the medulla, which sends messages to increase the rate and depth of breathing. Very soon the level of carbon dioxide is back to normal, but at the expense of a higher respiratory rate.
So back to Jonathan, he is mildly hypoxic, shown by low oxygen saturations, and has an increased respiratory rate at 28/min (normally less than 20/min). His lungs are not doing either of the things they are meant to do properly, take in oxygen or get rid of carbon dioxide. Oxygen saturation and respiratory rate are therefore the most important things to look at if you are worried about lung function. If they are normal, it’s likely that the lungs are working normally.
The next thing is chest expansion. What makes the chest wall move outwards when we breathe in? In healthy people sitting quietly, the chest wall hardly moves at all, because breathing is done by the diaphragm, acting like a bellows. Next time you are sitting next to someone in a lecture or watching television, look at their chest and you will see what I mean (I suggest you take care not to make it too obvious – sometimes scientific enquiry can be misinterpreted). When the diaphragm contracts it makes the abdominal wall move outwards. Jonathan’s chest was very obviously moving with each breath, although you could easily see that the left side moved less than the right – it is always the abnormal side which moves less. The chest moves outwards because the ribs are being pulled upwards by intercostal muscles pulling the ribs together. The whole rib cage moves upwards like a venetian blind being opened. The reason the ribs then move outwards is because of the way they are joined to the spine and sternum.
bucket handles must move outwards as they are lifted up
Like a bucket handle – if you pull it up it must move outwards. The outward movement increases the volume of the thoracic cavity and pulls air into the lungs. Jonathan is consciously stopping his left chest wall expanding – the inflamed pleural surfaces are rubbing together with each breath making it very painful.
I hope it’s clear that you can find an awful lot about a patient’s lung function without touching them. But all our doctors wear stethescopes around their necks, and it would be a shame not to use them. Before using the tubes, we can do useful things with our hands.
all junior doctors in our hospital wear a stethescope around their neck – it is a badge of office
First of all percussion. This is pretty straightforward. It takes a bit of practice but it is certainly useful to tell if there is a pleural effusion or consolidation. In Jonathan, who is thin, it was quite easy to tell that the percussion note was duller over his left lower chest at the back.
Then there is tactile vocal fremitus. Most non-respiratory doctors and students I have met seem to think this is something that is only worth doing in exams, and is an inferior test to listening with a stethoscope. This is not true. Testing tactile vocal fremitus involves putting your hands on the surface of the chest and asking the patient to say something and feeling whether the vibration can be felt with the palm or edge of the hand. In the UK doctors usually ask patients to say “ninety-nine”. Why ninety-nine? It comes from German physicians who realised that they needed the patients to say something with lots of low frequency sounds – “neunundneunzig” – the German for ninety-nine works very well. Ninety-nine in English doesn’t. I tried for a while getting patients to say “doom and gloom” but then one of the nurses took me aside and started asking about whether there was something I needed to talk about. At the suggestion of my wife, I am now trying “blue balloon” – seems to work pretty well. Why do we need a low frequency to feel the vibrations through the chest? I suggest you try an experiment – ask a good friend to you put your hands over his/her chest while she/he hums a note, starting low and slowly rising. If your friend is thin, the low notes will be easy to feel, but the buzzing feeling will disappear as soon as the note gets a bit higher. The frequency of a deep voice is about 150 cycles/second or 150Hz (middle C is 260Hz). The wavelength of that sound can be easily estimated – sound travels about 300 metres/second in air so that sound wave is about 2 metres long (of course you remember from school physics that the wavelength is equal to speed/frequency). When I calculated this I was surprised – I always thought that the wavelength of sound waves was tiny. In normal lung, these large wavelength noises are transmitted very easily, but when the note gets above 200Hz there is rapid attenuation, and at 2000Hz almost no sound gets through at all. That, and the fact that it is more difficult for hands to sense higher frequencies, mean that tactile vocal fremitus is better felt with lower frequencies.
low frequency sounds are transmitted through normal lung easily, but high frequency sounds are attenuated and not transmitted
These large, low frequency waves don’t get absorbed much by normal lung, but will be blocked if there is an air/fluid interface such as a pleural effusion. That means that tactile vocal fremitus is reduced or absent when there is fluid in the pleural space and can in fact be very useful for detecting quite small effusions. It is less useful in obese people because the fat overlying the chest wall is essentially fluid (see previous post about chest pain and haggis), and has a similar effect to pleural fluid. If you read textbooks about lung examination there is the suggestion that tactile vocal fremitus is increased over consolidated lung, such as in Jonathan’s case when he had a pneumonia. When we examined him he clearly had reduced tactile vocal fremitus at the bottom of his left lung, no doubt because the inflammation was associated with a small amount of fluid in the pleural space. I am not sure it is worth testing for fremitus anywhere apart from the lung bases, as it is only really good for looking for pleural effusions – but it is very good for doing that and should be routinely tested. If you disagree with this let me know – happy to host a discussion.
Next we come to vocal resonance. Listening over the chest wall while the patient says something. I usually ask them to say “Mary had a little lamb”. If you have a stethoscope try listening to a friend’s chest while they say this. If they have normal lungs it will sound strange – really muffled – more like mooohoodloodleloom. If you put the stethoscope on their forehead it will not be distorted. Why does normal voice become distorted when heard with the stethoscope applied to the chest in people with normal lungs? It is because the high frequency bits of the voice are lost. Linguists call the high frequency sounds plosives – the sounds we make with our voices which involve our tongue stopping the flow of air to create t or d or c sounds. These are high frequency, well over 200Hz and are filtered out when travelling through normal lung. Why does normal lung filter out higher frequencies – that is more difficult, and needs an understanding of the physics of sound transmission through foams. If you want to read more I suggest you start with:
The left lower lobe of Jonathan’s lung was consolidated – filled with pus – fluid and neutrophils. Angry neutrophils making bleach and causing a lot of collateral damage to lung tissue and blood vessels. The hypochlorite bleach will rapidly oxidise haemoglobin to methaemoglobin (see long lie and salami below) to produce the characteristic rusty-red sputum he was coughing up. Because the lung is no longer like a foam, containing little air, high frequency sound travels much more easily and “Mary had a little lamb” is no longer distorted as in normal lung. This phenomenon, where voice is heard faithfully over consolidated lung is also known as bronchophony.
Similarly normal breath sounds are altered over consolidated lung. When our lungs are healthy, and we are at rest, air flows into our lungs in a laminar fashion, and makes very little noise.
laminar, quiet flow
When the speed of air increases beyond a critical value, the airflow becomes turbulent. This makes it noisy. Somewhat like when water is run into a bathtub – slow flow gives a clear, almost noiseless flow. When the tap is turned on more the water comes out in a noisy, bubbly flow. But turbulence in airways is useful – it mixes up gas more to improve delivery of oxygen and removal of carbon dioxide. Why not have turbulent breathing all the time? – well for one we would not like to sound like Darth Vader all the time, and it probably would not be the best thing for hunter-gatherer man when staying quiet was probably quite important for survival.
turbulent, noisy flow
When we are running away from the hungry tiger a bit of noisy breathless breathing is probably not too much of a problem if it gives us better lung function. Turbulence causes a noise which has a hissing quality and has a whole range of frequencies from low to high – white noise. In normal lung the high frequencies are filtered out leaving a more muted sound (?pink noise) when heard on the chest wall – this is commonly described as vesicular breath sounds. When the high frequencies are also transmitted in consolidated lung the sound is harsher – bronchial breathing. If you want to hear what bronchial breathing sounds like put a stethoscope over that friend’s forehead while they take a deep breath in and out – the sound is not as loud as over the trachea but is not distorted. Whispering pectoriloquy is useful to confirm consolidation. We asked Jonathan to whisper “ninety-nine”. Whispered voice does not have any low frequency so cannot be heard well over normal lung. It could be heard clearly over clearly over his consolidated lung.
I could talk about other lung sounds such as crackles and wheezes, but this post has already become too long – maybe another time.
The next day Jonathan was much better, we grew a pneumococcus from his blood culture and he went home after two days on oral penicillin-based antibiotics.
The food link this week is scones.
scones my wife made last week with sour milk
I love scones, especially with jam and clotted cream and a nice cup of tea. They are traditionally made with sour milk and bicarbonate to make them rise in the oven. Milk goes sour when the lactose sugars are converted to lactic acid by lactobacilli. This lactic acid reacts at high temperature with the bicarbonate to make carbon dioxide gas that puffs up the scone to make it light and fluffy. This is the reverse of the reaction in our body when too much carbon dioxide turns into bicarbonate and acid (respiratory acidosis).
A few weeks ago we admitted Sylvia. She had been feeling unwell for a some days, with fever and episodes of uncontrollable shaking. What really alarmed her was when she turned yellow, and then developed abdominal pain and vomited a couple of times. Sylvia is forty eight, and had always been healthy. She rowed with the local club twice a week and kept her weight under control. For the past year or so she noticed that she got pains in her abdomen after eating fatty foods such as fish and chips, and now avoided foods like that. It was her son who noticed she was yellow – he then phoned Sylvia’s GP. The duty doctor came and confirmed that she was indeed jaundiced, feverish and unwell, and persuaded her that she needed to go to hospital.
these people look jaundiced – in fact they are not – the whites of their eyes are white – this image is copyrighted but Wiki thinks its ok to use it so I’m hoping its ok for me to do so
We did all the usual things for someone with suspected biliary sepsis. Took blood cultures and routine blood tests. These tests confirmed she had elevated bilirubin and transaminases, increased alkaline phosphatase, as well as a high neutrophil count and CRP. We gave her intravenous fluids, anti-emetics and antibiotics. We were able to get an ultrasound within a couple of hours. Ultrasound is really good at looking at the liver and biliary system in jaundiced patients – better than CT.
The ultrasound showed that Sylvia had a gallbladder full of gallstones and her common bile duct was dilated. It seemed likely that she had a stone blocking the flow of bile from the liver to the duodenum, and that the stagnant bile had become infected with bacteria causing ascending cholangitis and liver inflammation.
bile is made in the liver – it is stored in the gallbladder and squirted into the small intestine after a fatty meal, along with pancreatic secretions – Sylvia’s gallstone was impacted in the sphincter of Oddi
The questions I want to answer this week are:
Why did Sylvia turn yellow?
What are gallstones made of and why do they form?
Why is there an increase in alkaline phosphatase in the bloodstream when the biliary system is obstructed?
The last of those questions is the hardest, but I will make some conjectures, and hope that others who know more about it will correct me.
Jaundice is fairly straightforward. It is caused by the accumulation of bilirubin, which is a greenish-yellow colour. Bilirubin is made from haem, the central working part of myoglobin, but most bilirubin comes from the breakdown of haem in the haemoglobin from red blood cells.
So why is haemoglobin red? It is not only – or even mainly – the iron that makes haem red, but the porphorin ring that surrounds the iron. This large ring is made of four smaller pyrrole rings.
Pyrroles are often coloured – the dye indigo has two pyrrole rings and red acrylic paint often contains the dye pyrrole-red. Melanin, the pigment that gives skin and hair its colour is brown or black or red because it has a related structure – indole – a pyrrole stuck to a benzene ring. Without this pyrrole ring we would be like albinos, but without pink skin and eyes. As we shall see, we would also produce beige poo and colourless urine.
this is a pyrrole – all pyrroles are highly coloured, and are in most of the things which make people the colour they are
Red blood cells last on average 120 days before they wear out and have to be recycled. As we have five litres of blood and in each litre there is at least 120 grammes of haemoglobin, it means we have to make, and break down, five grammes a day. About a teaspoonful. This all happens inside macrophages in the spleen and liver, as they try to salvage useful parts of the red cells to make new things.
Macrophages are similar to neutrophils but slightly smarter. Some are good at host defence: they come to the scene after the neutrophils have done their job, do the forensics, and find out what the germs are made of. They then tell the lymphocytes how to make antibodies to protect us in the future.
Other macrophages are good at clearing up the mess, breaking down dead tissue and making things neat and clean. The spleen and liver macrophages that deal with old red cells are the latter kind. When they take red cells apart, they separate haem from globin, and then take the iron out of the haem. Next, the porphorin ring is broken to form bilirubin – its molecule of linked pyrrole rings looks like four beach huts in a row.
haem is broken down by the enzyme haem oxygenase to form bilirubin
The bilirubin is then thrown away (I don’t know why it can’t be re-used). It attaches to albumin and becomes water soluble after conjugating in the liver with a sugary molecule called glucuronate. The conjugated bilirubin is then transported into the biliary system, stored in the gallbladder, and squirted into the duodenum when we eat fatty food. In the bowel, it is converted to urobilinogen, which makes poo brown. Some is reabsorbed and excreted in urine, which makes urine yellow.
Sylvia had noticed that her poo had changed colour – it had become putty-coloured – and her urine had become much darker. That is because conjugated bilirubin was not getting into her intestines and instead some had leaked into her bloodstream and was appearing in her urine. The whites of her eyes and skin had become yellow because of the very high level of conjugated bilirubin in her blood.
Now to the gallstones. In Western countries they are most commonly made of cholesterol. I have already talked about the importance of cholesterol in keeping cell membranes rigid and non-leaky. Cholesterol is made in large amounts by the liver and transported to other cells gift-wrapped as LDL cholesterol. It is also used to make bile salts. It only needs a minor modification of the basic chemical structure of cholesterol to make bile salts – the main component in bile which makes it work – the main function of bile salts is to help us digest dietary fat.
structure of cholesterol
Bile salts are detergents. Just like washing-up liquid, they emulsify fats, breaking large fat globules into smaller micelles which do not stick to each other, or anything else. Cholesterol is essentially very insoluble in water, or hydrophobic. When an organic acid group is added – to make bile salts – it gains a hydrophilic or water-attracting group. This is just like soap – a long lipid chain with an organic acid group at the end. The hydrophobic lipid part is embedded in the tiny fat globule and the hydrophilic groups stick outside in the watery medium of the intestinal contents.
bile acids are a minor modification of cholesterol – hydrophilic OH and COOH groups are added to make it into a detergent
As well as bile salts, the liver secretes phospholipids into bile – principally phosphatidylcholine. This, with cholesterol, is the main stuff cell membranes are made of. It is also an emulsifying agent, and is used extensively in the food industry to keep fats in suspension.
So, when we eat fat, the small intestine detects it and releases the hormone cholecystokinin (CCK). This causes the gallbladder to contract to send bile into the duodenum, and also makes the pancreas release enzymes, including lipase. CCK also has some very interesting effects on our brains, making us feel less hungry, and curiously, opposing the effects of opiate drugs. The bile and pancreatic secretions are both delivered into the duodenum through the same tube – see diagram. The detergent bile salts, emulsifying phospholipid and pancreatic lipase are designed to work together to digest fat.
the hydrophilic parts of cholesterol are all on one side of the molecule – this allow it to act as a detergent – it works well to disperse fat into small globules so that pancreatic lipase can work on it and break it down to fatty acids and monoglyceride
The bile breaks it up into tiny globules and the lipase breaks triglyceride into fatty acids and monoglyceride. These are transported across the intestinal mucosal cell membrane and then the triglyceride is put back together again. Seems a daft system, but it clearly works. The triglyceride is taken away from the intestine not in the portal blood, like most other substances absorbed by the gut, but in the lymphatic system in the form of chylomicrons – small fatty globules covered by a layer of phospholipid. The chylomicrons travel up the lymphatic vessels to emerge into the circulation just below our left clavicle. This means that it avoids passing straight away through the liver. If blood is taken soon after a fatty meal and centrifuged, it will have a milky appearance because of the large amount of chylomicrons.
So why do gallstones form? As well as bile salts, unmodified cholesterol is also secreted into bile. It is only kept soluble by the bile salts and phospholipid – the ratio of cholesterol to bile salts and phospholipid is therefore important. When it goes wrong, cholesterol precipitates out and forms solid stones like those that caused all Sylvia’s recent problems – a bit like when I try to make mayonnaise and it curdles – the fat becomes un-emulsified and separates out.
In some parts of the world, gallstones are made from bile pigment – bilirubin. This can be due to increased red blood cell breakdown resulting from abnormalities such as sickle cell disease, but it’s also common in East Asia, for reasons which are not clear. A good, comprehensive analysis of why gallstones of all types form can be found at:
Now the alkaline phosphatase. Bile is alkaline. Alkaline phosphatase is an enzyme that removes phosphate groups in alkaline conditions. When bile flow is blocked – by stones, as in Sylvia’s case, or for any other reason – the liver makes much more alkaline phosphatase, some of which appears in the blood. Bones use a similar alkaline phosphatase to rearrange phosphate groups to make hydroxyapatite – the hard stuff bone is made from. Most of the alkaline phosphatase normally in our blood is the bone sort.
There is plenty of information about whether alkaline phosphatase is from bone or liver, but very little I can find that suggests why the liver should make this enzyme when the biliary system is blocked. I think a clue here is that neutrophils also have alkaline phosphatase in their granules, which help kill bacteria. This is in addition to the myeloperoxidase and esterase mentioned in earlier posts. Why should a phosphatase be damaging to bacteria? The answer may well involve techoic acid, an important reinforcing molecule in some gram-positive bacteria.
cartoon of gram positive bacterial cell wall – techoic acid is thought to inhibit the action of lysozyme and protect the peptidoglycan from being broken down – techoic acid contains phosphate groups which may be removed by alkaline phosphatase
In these microbes, techoic acid strengthens the peptidoglycan cell wall and inhibits the action of lysozyme, yet another enzyme made by neutrophils. Lysozyme is designed to break some of the sugar-sugar bonds in peptidoglycan – so getting rid of the techoic acid would surely be helpful. We know that alkaline phosphatase is effective in breaking up techoic acid – see, for instance:
Maybe the liver is producing this anti-bacterial enzyme to help prevent infection when the flow of bile slows down. Unfortunately, this won’t work with gram negative organisms such as E.coli because they don’t make techoic acid – and it was a gram negative bacterium we grew from Sylvia’s blood culture – the germ that was causing her ascending cholangitis.
Soon after she came in, Sylvia had an ERCP – an endoscopic retrograde cholangio-pancreatoscopy. The endoscopist managed to remove the impacted gallstone from her ampulla of Vater, and she quickly recovered. A week later she had her gallbladder removed, including the stones, and now is quite well – she can even eat fish and chips.
Now to the food link: mayonnaise.
French mayonnaise – made with olive oil and mustard
This is an emulsion of lipid, such as olive oil, in an aqueous (watery) medium – vinegar or lemon juice. The emulsifying agent is raw egg yolk, which – just like bile – contains lots of phospholipid – phophatidylcholine and is also rich in cholesterol. In France, mustard is also added. Mustard seed contains mucilage, a gooey stuff that some plants produce made of sugar polymers. (Thanks again to Harold McGee). These also act as emulsifiers and further thicken the mayonnaise, and give it a better taste. When I make cheese sauce, I use a sugar polymer (flour) to achieve a stable emulsion between butter and milk – no doubt the phospholipid in the milk helps too. It seems likely that in the small intestine, the mucus from the stomach, a similar gloopy sugar polymer, probably has a similar effect to mustard seed mucilage to help with fat emulsification.
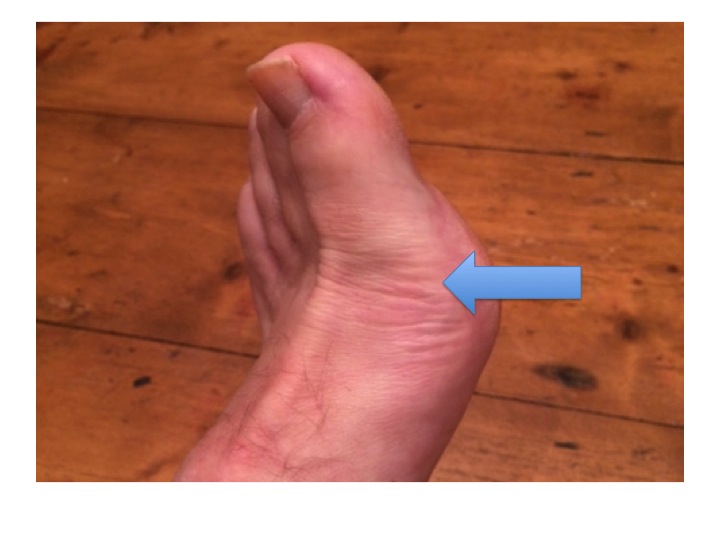


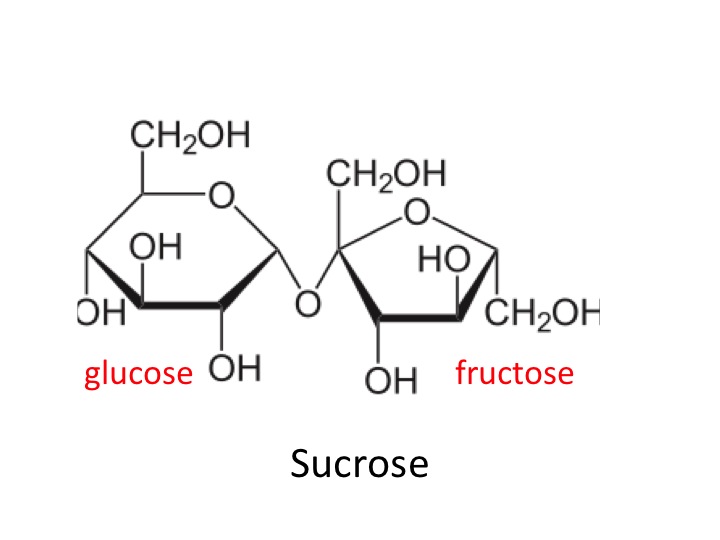

 sucrose and high fructose corn-syrup, is much more important than saturated fat in causing obesity, diabetes and vascular disease. His videos and book are worth looking at:
sucrose and high fructose corn-syrup, is much more important than saturated fat in causing obesity, diabetes and vascular disease. His videos and book are worth looking at:
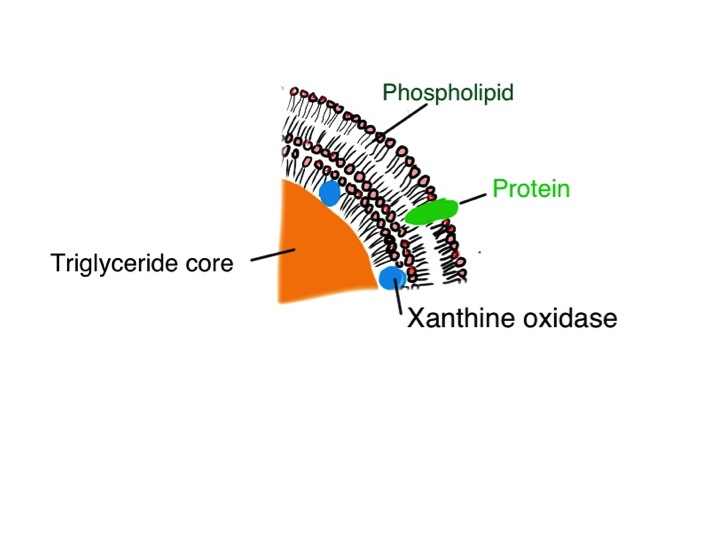
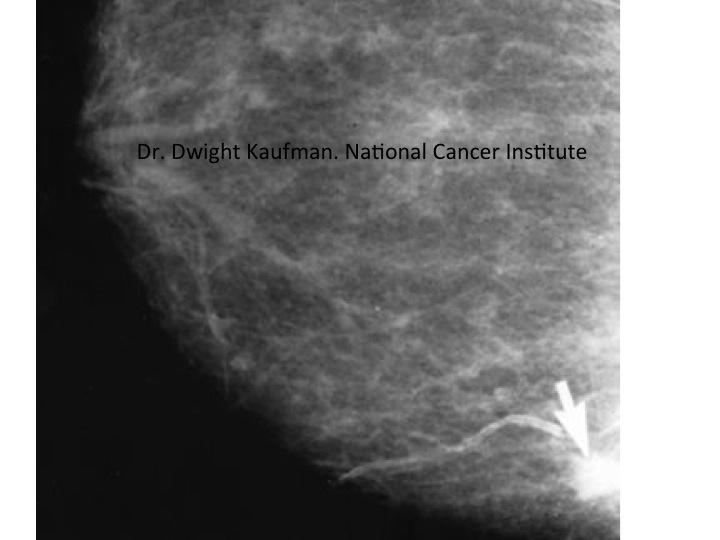






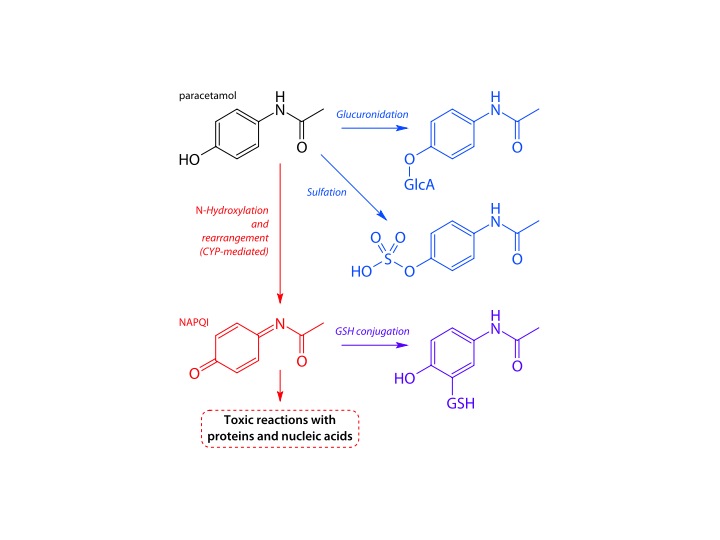





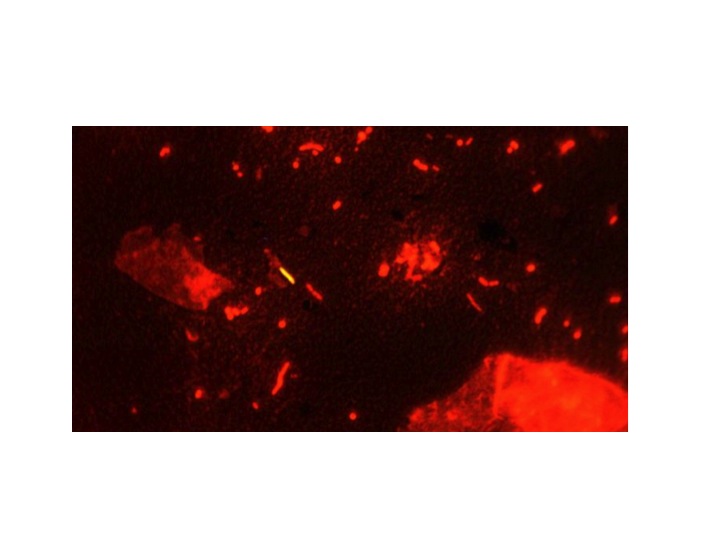

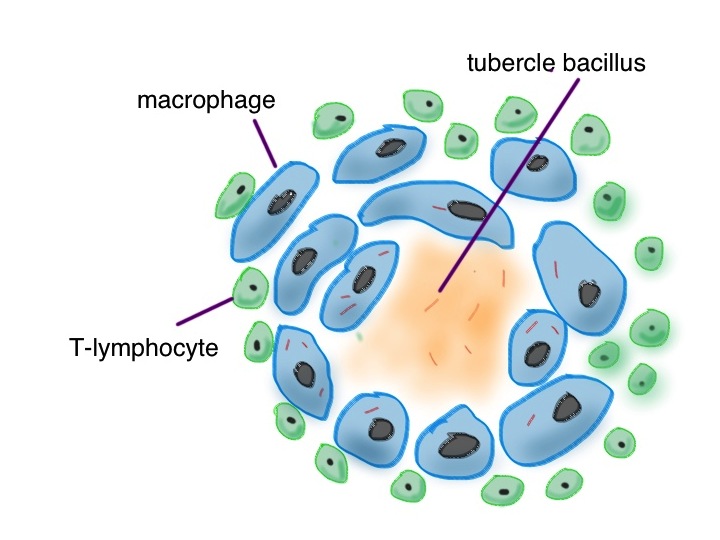

![Campylobacter are helical, like H pylori - it helps them swim through the mucus layer and invade into the intestinal epithelium By De Wood, Pooley, USDA, ARS, EMU. [Public domain], via Wikimedia Commons](https://wardroundstuff.com/wp-content/uploads/2013/12/slide13.jpg)