We admit a lot of elderly patients who have fallen over. There are a whole range of problems which cause falls, one of which is postural, or orthostatic hypotension. Emily is 78 and lives alone in a bungalow. Her husband died three years ago, but her daughter calls in to help most days. She was fairly well until she went to her family doctor’s surgery last week and had her blood pressure measured. The result was not good – 186/88mmHg.
She first developed high blood pressure when she was in her fifties, and it had crept up since. She was already taking ramipril (an ACE inhibitor) and indapamide (a thiazide diuretic). She had tried amlodipine, but that gave her swollen ankles, which she hated. Her GP decided to try her on doxazosin. This is an alpha-blocker that lowers blood pressure by opening up small arteries. The morning of the day of admission she got up and took her tablets. Then she had a bath, but when she got out felt awful, and faint, and fell to the floor. She lay on the floor for a few minutes and tried to get up, but felt very dizzy and had to lie down again. She phoned her next-door neighbour on her mobile phone. The neighbour came round and helped Emily to her feet, but she went wobbly and could not stand, ending up on the floor again. Her neighbour became really worried and called for the ambulance. When the ambulance paramedics arrived they found that when they tried to stand Emily up, her blood pressure dropped from 160mmHg systolic to 90mmHg. They brought her in to see us.
There is a popular notion that all antihypertensive drugs cause postural hypotension, but this is not the case. As we will see, beta-blockers can even be used to treat this condition.
What is happening with the circulation to cause Emily’s blood pressure to drop when she stands up?
standard clinical rubber glove
The whole explanation is made much easier with a standard hospital rubber glove (the stretchier the better) and a sink, and a supply of water. First we fill the glove up with the right amount of water.
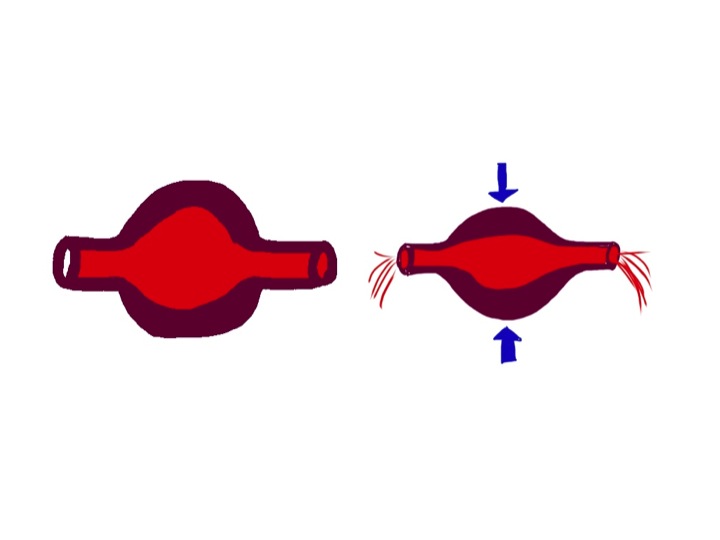
glove about half filled with water – laid flat – notice the thumb is filled with water – this represents cardiac filling pressure
The water in the glove represents blood in the venous side of the circulation. The arterial side is much lower volume, and is very little affected by changes in pressure due to posture.
When we stand up the amount of fluid in the veins of the legs and abdomen increases, due to increase in hydrostatic pressure stretching the rubbery material. Veins are thin-walled and rubbery, and will naturally distend if the pressure inside is increased. If this happens, the thumb empties, this represents the filling pressure to the heart. If cardiac filling pressure drops, cardiac output drops and blood pressure falls.
no venous squeeze due to failure of the autonomic system, due to brain stem or peripheral sympathetic nerve action means no cardiac filling pressure – the thumb is empty
This will result in a fall in systemic arterial blood pressure – postural hypotension.
But this does not happen in healthy, young people. We have autonomic reflexes which cause constriction of veins before we stand up, so that filling pressure to the heart is maintained. In the glove model this is represented by my hand squeezing the fingers and main hand of the glove.
in healthy individuals the autonomic nervous system provides a squeeze of abdominal and leg veins to maintain filling pressure of the heart – notice the thumb is now full of water
There are two main causes for postural hypotension – failure of autonomic squeeze of the veins, and insufficient filling of the venous compartment.
Let’s take the first of these – not enough squeeze. When we move from lying down to standing up, our brain knows what is about to happen. As we move our limbs the proprioceptors in our joints tell our brain what is about to happen and our conscious mind probably also has a role. Messages are sent to the control centre (Houston – prepare for lift-off) in the medulla. Sympathetic nerves send messages down to the veins to tell them to constrict. It is interesting that human intra-abdominal mesenteric veins are particularly richly supplied with sympathetic nerve endings – presumably because we are one of the few animals where standing is such a problem. In animals such as horses and dogs, moving from lying to standing does not involve a capacious venous network to be subjected to large pressure changes – their legs have very little venous blood and their abdomen and contents is on the same level as the heart.
It is also not surprising that if you take a healthy, young person and strap them to a tilt-table, and suddenly move them from horizontal to vertical, they will experience an impressive drop in blood pressure. If they are lying on a bed and stand up – blood pressure hardly changes because the brain-stem prepares us for the change in posture with messages to our abdominal and leg veins to constrict.
As we get older, everything starts to go wrong (see last week’s post). Autonomic reflexes become impaired. Patients with Parkinson’s disease are particularly prone to develop postural hypotension because of impaired brain stem autonomic reflexes – this used to be called Shy-Drager syndrome when I was young – it is now know as multi-system atrophy. Some drugs such as antidepressants and methydopa inhibit brain stem sympathetic output and predispose to this condition.
The sympathetic nerves end up on the outside (adventitia) of veins and release noradrenaline which causes constriction of venous smooth muscle by stimulating alpha receptors. Emily had been given an alpha-blocker to reduce constriction of her arterioles and thereby reduce blood pressure. In her it reduced the ability of veins to constrict when standing up by blocking venous alpha receptors.
Some patients have problems with the autonomic nerves. Diabetics and alcoholics are prone to develop autonomic neuropathy which may result in postural hypotension due to inability of veins to constrict.
Temperature also plays a part in venous constriction. Most of us have noticed occasionally when getting out of a hot bath or sauna feeling dizzy for a few seconds, we hold onto the edge of the bath then things improve. What has happened is that the hot bath water has caused venous dilatation and the normal reflexes have not been enough to maintain cardiac filling pressure. Cardiac output drops, baroreceptors panic (Houston, we have a problem). Houston responds quickly by sending a stronger sympathetic signal to the veins and to the heart to increase cardiac output and the problem is soon sorted. But of course preventing the problem is much better than reacting when it has happened.
The second main cause of postural hypotension is insufficient fluid in the circulation.
to represent fluid loss – some water is poured out of the glove
In Emily’s case this may have been partly due to the thiazide diuretic she was taking for her blood pressure. Patients on loop diuretics such as furosemide are even more prone to fluid depletion. Sepsis causes problems with a postural blood pressure drop because of fluid shifts out of the circulation into interstitial spaces, and because fever causes venous dilatation as in the hot bath above. Jonathan, who had a lobar pneumonia a few weeks ago, may well have collapsed in his GP’s surgery because of a postural blood pressure problem.
even with reduced volume – when lying flat the thumb is still filled with water and blood pressure is maintained
Blood loss from trauma or intestinal bleeding may often not result in blood pressure drop until the patient stands up – it is far easier to maintain filling pressure when lying down and not having to squeeze the veins hard.
with reduced volume, squeezing the legs and abdomen no longer maintains filling of the thumb – cardiac filling reduces and blood pressure drops
Sorting out Emily’s postural hypotension was straightforward. We gave her two litres of intravenous saline and stopped her doxazosin, and diuretics. We had a plan to start her on verapamil or diltiazem when she could stand up without her blood pressure dropping – which it did the next day.
Do ACE inhibitors/ARBs, beta blockers or calcium channel blockers cause postural hypotension?
The answer is to begin with, no and no.
ACE inhibitors can cause a postural drop when they are first started. This is because angiotensin does have an effect on venous constriction, but only transiently when it is increased or decreased. If angiotensin II is infused into a hand vein it will constrict, but only for a few hours at most. Similarly if angiotensin II is withdrawn, the vein will dilate, but only briefly. This means that it is prudent to warn patients that they may develop postural symptoms following the first dose – and it is a good idea to take it when lying down before going to bed. The effect of angiotensin II on arterioles is long-lasting, which is why these drugs are useful in treating arterial hypertension.
Beta blockers reduce blood pressure by mechanisms which are not completely understood, but is likely to be a combination of reduced cardiac output and inhibition of renin release from the kidney. There are beta2 receptors in arteries and veins, but these cause vascular dilatation. Beta blockers can help veins constrict, which is why they are sometimes used to treat postural hypotension. Having said that, if there is a postural drop in blood pressure, the reflex mechanisms set in train by baroreceptors involve increased sympathetic stimulation to the heart via beta1 receptors, increasing force and rate of contraction. Beta blockers will block this response and may therefore impair the recovery from an episode of postural hypotension, but they will not cause it.
Calcium channel blockers have an effect on arterioles, not on veins. Calcium channels are important in maintaining arterial tone, but not venous tone. Venous tone depends almost entirely on sympathetic stimulation. Therefore, at least in theory, drugs such as verapamil, diltiazem and amlodipine will not cause a postural drop. But of course if there is a postural drop, lower initial blood pressure caused by these agents may make the episode worse.
Sorting out Emily’s orthostatic hypotension was fairly straightforward. What about when it is caused by age-related impairment of brain stem function, or by irreversible peripheral autonomic neuropathy? It can be a real problem.
overfilling the glove means that even with no squeeze to the veins the thumb remains full of water when the glove is upright – overfilling the circulation with fludrocortisone can help prevent postural hypotension
One approach is to use fludrocortisone. This is a synthetic analogue of aldosterone. Made from cholesterol (again!) this hormone is made in the adrenal cortex, in different cells which make cortisol from cholesterol. The function of aldosterone is to regulate how much salt and water is excreted from our kidneys. Aldosterone helps the reabsorption of more salt from the distal convoluted tubule and will therefore increase circulating volume of blood. This will have the opposite effect to diuretics and overfill the circulation and help keep filling pressure at heart level adequate on standing up. Another drug which is sometimes used is midodrine, an alpha receptor agonist, helping constrict veins and maintain cardiac filling when upright. The drug does not have a licence for this indication in the UK.
We have made a not very good video showing how postural hypotension works – my first attempt at youtube – thanks for your help Steph. We will try to make a better one soon and update this post. The video is at:
The food link this week was not obvious when I started, but of course it has to be liquorice.
liquorice has been popular for a long time – it is made from the root of the liquorice plant
Liquorice contains glycyrrhizin which has a chemical structure similar to steroids such as cholesterol and aldosterone (made from cholesterol – again!). It has long been known to have a mineralocorticoid effect. This is not because it acts directly in the kidney to stimulate aldosterone receptors. Instead it prevents the conversion of cortisol to cortisone. Cortisone, synthesized by the adrenal glands, is converted to cortisol in the kidney, then back to cortisone, cortisol has more mineralocorticosteroid effects than cortisone – full details are in the NEJM paper:
This is the third in a series of rigors. Some doctors call them chills, but I think of a rigor as a really uncontrollable shaking episode. I remember when I was a junior surgical house officer I was looking after patients who had undergone prostate surgery. From the nursing station at one end of the ward, I would hear a bed shaking at the other end. The beds in those days had metal frames and really clanked loudly when someone had a rigor – which on the urology ward was due to gram-negative septicaemia.
“Could you please draw up some gentamicin and ampicillin?” I would ask the nurse as I walked down the ward, armed with blood culture bottles to see an elderly man looking very alarmed, shaky and unwell. I think these days we are more careful to make sure our patients do not have infected urine before operating on them.
Earlier this week we admitted Jonathan. A very fit 36 year-old man who had never been in hospital before, apart from when he had an arthroscopy for a damaged knee following a football injury. It started with him feeling off-colour, off his food, and generally aching all over. He thought he had the ‘flu. He woke up at four in the morning to find it really hurt on the left side of his chest when he tried to breathe deeply. He had never experienced pain like it before, even after the arthroscopy. Then he started shaking uncontrollably. He then began coughing, and that was even more painful. Then he coughed up some brownish-reddish, rusty-coloured dark phlegm. He got up and took some paracetamol, went back to bed, but could not sleep. He decided he could not go to work and went instead to his GPs surgery. He had another rigor while in the waiting room, stood up and collapsed to the floor. The practice nurse found a couch so he could lie down, and got the GP to see him straight away. He was with us a couple of hours later. By the time we got to see him he had had a chest X-ray which showed a left lower lobar pneumonia.
Pyelonephritis, ascending cholangitis and lobar pneumonia are the diseases which commonly cause severe rigors. Malaria is another we see less commonly in our part of the UK, but can certainly also produce impressive rigors. To get rigors you need to have a flood of microbes released over a short period of time, which happens if you have an organ such as the kidney, liver or lung full of germs. Other infections cause fever, but less commonly rigors.
The reason for shaking with rigors is that our body is trying to increase our temperature quickly. The mechanism is similar to shivering when we are cold – using muscle contraction to generate heat. Substances on the bacterial cell wall are recognised by Toll-like receptors on macrophages. This is part of the innate immune system which is designed to respond to a whole variety of germs. Toll-like receptors are on a range of white blood cells, intestinal epithelial cells, respiratory epithelial cells and vascular endothelium – all the places we need to keep a close eye on where the enemy is likely to invade. Lipopolysaccharide, the coating of gram-negative germs is the classic trigger. Techoic acid on the surface of gram positive organisms (see last week’s post) is another. These receptors can even recognise DNA sequences only found in bacterial but not in mammalian cells. When macrophages are alerted to the presence of bacteria, they generate cytokines such as interleukin 1 which circulate and are sensed by the hypothalamus in the brain. The hypothalamus is the part of the brain which regulates body temperature and coordinates the shaking response. When Jonathan started shaking he felt really cold, even though his body temperature was higher than normal. When we saw him in the emergency department he was wrapped in three blankets, even though it is always warm in there.
Why do we develop a fever with infections? There is some evidence that some bacteria such as Salmonella typhi, which causes typhoid fever, are less able to survive at 40 degrees than 37 degrees. Not sure it helps much with other germs.
The junior doctor took the history and examined Jonathan. He took blood cultures and prescribed intravenous co-amoxyclav and clarithromycin.
Making a diagnosis of lobar pneumonia is usually easy in hospital, when we have an X-ray to look at, although in the early stages the changes can be quite difficult to see. We are all taught as medical students what clinical signs to look for. I thought it would be useful to think about why they happen in patients with lung problems, and particularly pneumonia.
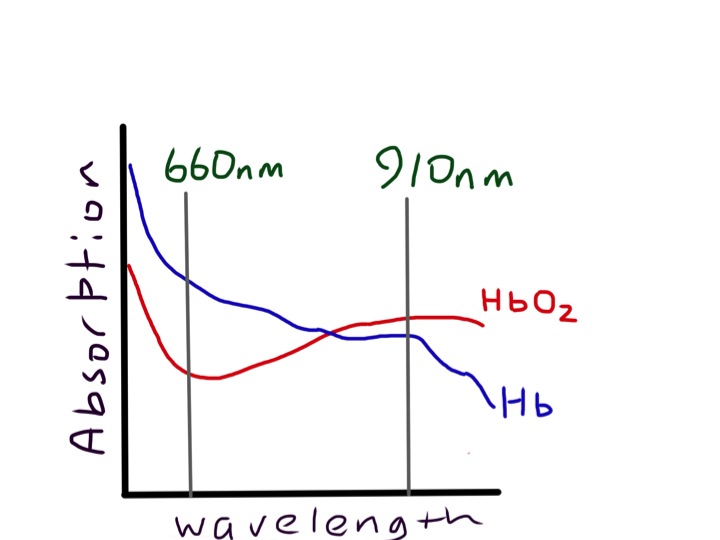
First it is worth thinking what the lungs are for. They do two main things: 1) get oxygen into our bloodstream from the air and 2) remove carbon dioxide from our blood so we can breathe it out. What happens when these vital functions don’t work properly? Normally our lungs are very good at getting oxygen into our bloodstream. Blood coming out of our lungs into the pulmonary vein and then into our main systemic arteries is usually saturated to 98%. We can measure this easily with a pulse oximeter. This is a wonderful device which has revolutionised our management of patients with respiratory conditions. It was invented in 1972 by Japanese engineers called Takuo Aoyagi and Michio Kishi. The way it works is that it shines two beams of light through the finger tip or earlobe. One beam is red light at wavelength about 610nm, the other is in the near infra-red at 910nm.
oxy and deoxy haemoglobin have different absorption characteristics, which makes them different colours – the sats meter measures absorption at two different wavelengths and compares them, but only in the pulsatile part of the signal
If you look at a pulse oximeter you can only see one red light, the infra-red is not visible.
Measurement of oxygen saturation relies on the absorbance of oxyhaemoglobin being different from deoxyhaemoglobin. We are talking about the colours of haem and pyrroles – again! Oxygenated arterial blood is well known to be redder than venous desaturated blood. The diagram shows that the absorbance spectrum of haemoglobin is very different at 610nm in the two types of haemoglobin. But at 900nm it is very similar. The machine compares the absorbance at the two wavelengths and can calculate the percent oxyhaemoglobin to deoxyhaemoglobin.
photo of pulse oximeter probe – interestingly it looks very differenet in the photo from real life because my mobile phone camera captures infra-red images which we cannot see with our eyes normally – try it yourself
If it just did that, we would find that the haemoglobin in a fingertip is normally about 70% saturated, because most of the blood is in veins, rather than arteries. The really clever thing about a pulse oximeter is that it analyses the ratio of oxy- to deoxyhaemoglobin just in the pulsatile part of the signal. As the pulsatile part is due to arterial filling of the fingertip, this gives arterial, rather than venous oxygen saturation. Jonathan’s arterial saturation was 91% before he was given oxygen therapy, when it rose to 98%. There is a common misconception that hypoxia will make you breathless. Chemoreceptors in the carotid body and brain stem can certainly respond to hypoxia, but only when it is very reduced. Flying at 30,000 feet in a jet plane is equivalent to being at 8,000feet altitude where the oxygen tension is reduced by about 20%. Passengers in jet planes will often have oxygen saturations around 92% but do not feel breathless. In fact hypoxia does not cause breathlessness unless it is really marked. Why is this? – The reason is probably because increasing the rate of breathing does not help. Without oxygen, if I asked Jonathan to breathe faster, firstly he would find it difficult because it was so painful, but even if he did, his oxygen saturation would not improve. This is because there is shunting of blood in the part of his lung affected by the pneumonia. In the inflamed part of the lung there is increased blood flow, but the air spaces are full of pus. There is consolidation. Any blood which flows through this part of the lung will not be oxygenated and will emerge into the pulmonary vein desaturated. Even if the rest of the lung is working perfectly, saturating the blood to 100%, there is no way to resaturate the desaturated blood which has travelled through the abnormal lung, so his sats meter will detect this and show a low reading. It looks like evolution has sussed this out and doesn’t bother to make us breathe harder – unless the oxygen saturation drops to the mid 80s when various tissues in our body start to complain and generate lactic acid.
Carbon dioxide is a different story. Every day we breathe out about one kilo or two pounds of carbon dioxide. Our lungs are not designed to get rid of CO2 completely. Jonathan’s blood gas analysis showed a normal pCO2 of 5KPa (normal range 3.5-6). We need a certain amount of carbon dioxide in our blood to keep the acidity normal. But most (80%) of the carbon dioxide is in the form of bicarbonate (HCO3), made from carbon dioxide by the enzyme carbonic anhydrase in red blood cells.
carbon dioxide is in equilibrium with acid and bicarbonate
If our lungs are not working, then carbon dioxide levels in the blood increase, bicarbonate increases, and hydrogen ion concentration increases, because of the balance between carbon dioxide and bicarbonate (see equation). We are all familiar with the reaction of bicarbonate and acid (vinegar or lemon juice) to produce carbon dioxide.
bicarbonate and vinegar
This is a reversible equilibrium reacation and in blood this can work the other way round so carbon dioxide makes acid and bicarbonate. This is known as respiratory acidosis. Unlike oxygen, our body is very sensitive to carbon dioxide levels. This is first detected in the carotid body, located at the division of the common carotid artery into internal and external branches, either side of the thyroid cartilage. This acts as an “early warning” signal, sending messages to the brain stem that
when mixed together produce lots of carbon dioxide
something is amiss and the lungs aren’t working properly. A few seconds later blood reaches the medulla in the brain stem, which is the central control mechanism for breathing, next to the vasomotor centre controlling the circulation. High carbon dioxide also stimulates the medulla, which sends messages to increase the rate and depth of breathing. Very soon the level of carbon dioxide is back to normal, but at the expense of a higher respiratory rate.
So back to Jonathan, he is mildly hypoxic, shown by low oxygen saturations, and has an increased respiratory rate at 28/min (normally less than 20/min). His lungs are not doing either of the things they are meant to do properly, take in oxygen or get rid of carbon dioxide. Oxygen saturation and respiratory rate are therefore the most important things to look at if you are worried about lung function. If they are normal, it’s likely that the lungs are working normally.
The next thing is chest expansion. What makes the chest wall move outwards when we breathe in? In healthy people sitting quietly, the chest wall hardly moves at all, because breathing is done by the diaphragm, acting like a bellows. Next time you are sitting next to someone in a lecture or watching television, look at their chest and you will see what I mean (I suggest you take care not to make it too obvious – sometimes scientific enquiry can be misinterpreted). When the diaphragm contracts it makes the abdominal wall move outwards. Jonathan’s chest was very obviously moving with each breath, although you could easily see that the left side moved less than the right – it is always the abnormal side which moves less. The chest moves outwards because the ribs are being pulled upwards by intercostal muscles pulling the ribs together. The whole rib cage moves upwards like a venetian blind being opened. The reason the ribs then move outwards is because of the way they are joined to the spine and sternum.
bucket handles must move outwards as they are lifted up
Like a bucket handle – if you pull it up it must move outwards. The outward movement increases the volume of the thoracic cavity and pulls air into the lungs. Jonathan is consciously stopping his left chest wall expanding – the inflamed pleural surfaces are rubbing together with each breath making it very painful.
I hope it’s clear that you can find an awful lot about a patient’s lung function without touching them. But all our doctors wear stethescopes around their necks, and it would be a shame not to use them. Before using the tubes, we can do useful things with our hands.
all junior doctors in our hospital wear a stethescope around their neck – it is a badge of office
First of all percussion. This is pretty straightforward. It takes a bit of practice but it is certainly useful to tell if there is a pleural effusion or consolidation. In Jonathan, who is thin, it was quite easy to tell that the percussion note was duller over his left lower chest at the back.
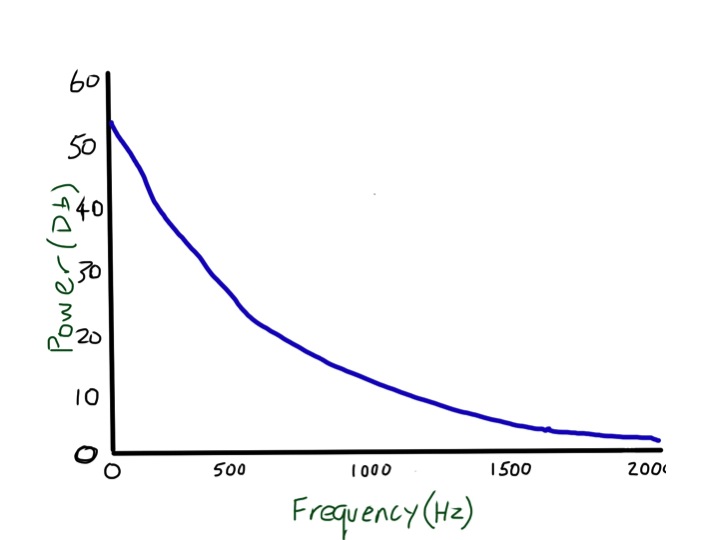
Then there is tactile vocal fremitus. Most non-respiratory doctors and students I have met seem to think this is something that is only worth doing in exams, and is an inferior test to listening with a stethoscope. This is not true. Testing tactile vocal fremitus involves putting your hands on the surface of the chest and asking the patient to say something and feeling whether the vibration can be felt with the palm or edge of the hand. In the UK doctors usually ask patients to say “ninety-nine”. Why ninety-nine? It comes from German physicians who realised that they needed the patients to say something with lots of low frequency sounds – “neunundneunzig” – the German for ninety-nine works very well. Ninety-nine in English doesn’t. I tried for a while getting patients to say “doom and gloom” but then one of the nurses took me aside and started asking about whether there was something I needed to talk about. At the suggestion of my wife, I am now trying “blue balloon” – seems to work pretty well. Why do we need a low frequency to feel the vibrations through the chest? I suggest you try an experiment – ask a good friend to you put your hands over his/her chest while she/he hums a note, starting low and slowly rising. If your friend is thin, the low notes will be easy to feel, but the buzzing feeling will disappear as soon as the note gets a bit higher. The frequency of a deep voice is about 150 cycles/second or 150Hz (middle C is 260Hz). The wavelength of that sound can be easily estimated – sound travels about 300 metres/second in air so that sound wave is about 2 metres long (of course you remember from school physics that the wavelength is equal to speed/frequency). When I calculated this I was surprised – I always thought that the wavelength of sound waves was tiny. In normal lung, these large wavelength noises are transmitted very easily, but when the note gets above 200Hz there is rapid attenuation, and at 2000Hz almost no sound gets through at all. That, and the fact that it is more difficult for hands to sense higher frequencies, mean that tactile vocal fremitus is better felt with lower frequencies.
low frequency sounds are transmitted through normal lung easily, but high frequency sounds are attenuated and not transmitted
These large, low frequency waves don’t get absorbed much by normal lung, but will be blocked if there is an air/fluid interface such as a pleural effusion. That means that tactile vocal fremitus is reduced or absent when there is fluid in the pleural space and can in fact be very useful for detecting quite small effusions. It is less useful in obese people because the fat overlying the chest wall is essentially fluid (see previous post about chest pain and haggis), and has a similar effect to pleural fluid. If you read textbooks about lung examination there is the suggestion that tactile vocal fremitus is increased over consolidated lung, such as in Jonathan’s case when he had a pneumonia. When we examined him he clearly had reduced tactile vocal fremitus at the bottom of his left lung, no doubt because the inflammation was associated with a small amount of fluid in the pleural space. I am not sure it is worth testing for fremitus anywhere apart from the lung bases, as it is only really good for looking for pleural effusions – but it is very good for doing that and should be routinely tested. If you disagree with this let me know – happy to host a discussion.
Next we come to vocal resonance. Listening over the chest wall while the patient says something. I usually ask them to say “Mary had a little lamb”. If you have a stethoscope try listening to a friend’s chest while they say this. If they have normal lungs it will sound strange – really muffled – more like mooohoodloodleloom. If you put the stethoscope on their forehead it will not be distorted. Why does normal voice become distorted when heard with the stethoscope applied to the chest in people with normal lungs? It is because the high frequency bits of the voice are lost. Linguists call the high frequency sounds plosives – the sounds we make with our voices which involve our tongue stopping the flow of air to create t or d or c sounds. These are high frequency, well over 200Hz and are filtered out when travelling through normal lung. Why does normal lung filter out higher frequencies – that is more difficult, and needs an understanding of the physics of sound transmission through foams. If you want to read more I suggest you start with:
The left lower lobe of Jonathan’s lung was consolidated – filled with pus – fluid and neutrophils. Angry neutrophils making bleach and causing a lot of collateral damage to lung tissue and blood vessels. The hypochlorite bleach will rapidly oxidise haemoglobin to methaemoglobin (see long lie and salami below) to produce the characteristic rusty-red sputum he was coughing up. Because the lung is no longer like a foam, containing little air, high frequency sound travels much more easily and “Mary had a little lamb” is no longer distorted as in normal lung. This phenomenon, where voice is heard faithfully over consolidated lung is also known as bronchophony.
Similarly normal breath sounds are altered over consolidated lung. When our lungs are healthy, and we are at rest, air flows into our lungs in a laminar fashion, and makes very little noise.
laminar, quiet flow
When the speed of air increases beyond a critical value, the airflow becomes turbulent. This makes it noisy. Somewhat like when water is run into a bathtub – slow flow gives a clear, almost noiseless flow. When the tap is turned on more the water comes out in a noisy, bubbly flow. But turbulence in airways is useful – it mixes up gas more to improve delivery of oxygen and removal of carbon dioxide. Why not have turbulent breathing all the time? – well for one we would not like to sound like Darth Vader all the time, and it probably would not be the best thing for hunter-gatherer man when staying quiet was probably quite important for survival.
turbulent, noisy flow
When we are running away from the hungry tiger a bit of noisy breathless breathing is probably not too much of a problem if it gives us better lung function. Turbulence causes a noise which has a hissing quality and has a whole range of frequencies from low to high – white noise. In normal lung the high frequencies are filtered out leaving a more muted sound (?pink noise) when heard on the chest wall – this is commonly described as vesicular breath sounds. When the high frequencies are also transmitted in consolidated lung the sound is harsher – bronchial breathing. If you want to hear what bronchial breathing sounds like put a stethoscope over that friend’s forehead while they take a deep breath in and out – the sound is not as loud as over the trachea but is not distorted. Whispering pectoriloquy is useful to confirm consolidation. We asked Jonathan to whisper “ninety-nine”. Whispered voice does not have any low frequency so cannot be heard well over normal lung. It could be heard clearly over clearly over his consolidated lung.
I could talk about other lung sounds such as crackles and wheezes, but this post has already become too long – maybe another time.
The next day Jonathan was much better, we grew a pneumococcus from his blood culture and he went home after two days on oral penicillin-based antibiotics.
The food link this week is scones.
scones my wife made last week with sour milk
I love scones, especially with jam and clotted cream and a nice cup of tea. They are traditionally made with sour milk and bicarbonate to make them rise in the oven. Milk goes sour when the lactose sugars are converted to lactic acid by lactobacilli. This lactic acid reacts at high temperature with the bicarbonate to make carbon dioxide gas that puffs up the scone to make it light and fluffy. This is the reverse of the reaction in our body when too much carbon dioxide turns into bicarbonate and acid (respiratory acidosis).
Heart failure is a very common reason to be admitted to our hospital. John, age 73, had a very typical story. He was well until about 3 years ago when out of the blue he had a major heart attack. Life has never been quite the same since. He has to take lots of tablets – aspirin, statins, beta blockers, ACE inhibitors and several fish oil capsules.
His wife worries about John a lot. She no longer makes him steak and kidney puddings and has stopped him smoking cigars when he goes out with his friends on a Saturday night. He has bought a dog and now takes much more exercise and drinks a lot less beer. But life wasn’t too bad until he noticed he was getting more short of breath when he took the dog for a walk a few weeks ago. Then he noticed ankles were swelling – his socks would leave deep rings around his ankles which disappeared in the morning. For the past week he found he could not lie flat without feeling short of breath.
Then the night he was brought into hospital he woke up gasping for breath. He was pale and sweaty and his wife got really worried and called the ambulance. By the time the paramedics got there he was still quite breathless and his ECG was abnormal, so they brought him in to hospital. He was given some nitroglycerin spray under his tongue- this helped a lot. In the emergency department he was given a dose of intravenous furosemide that made him “pee for Britain”.
We examined him on the admissions unit and found he still had some swelling in both his ankles which left dents when squeezed – bilateral pitting oedema- what was once known as cardiac dropsy. His neck veins were fuller than normal and he still had crackles at the base of his lungs when we listened. He had pulmonary oedema due to heart failure.
I really don’t like the term heart failure. You will know that I’m not one for using long Latin words but something like “impaired cardiac function” might be a less alarming label.
So why did John’s ankles swell up and why did he become breathless? When I ask students and junior doctors they usually say something about back pressure and right heart failure. Maybe it’s just in the UK, but I suspect there is a lot of confusion and misunderstanding about the physiology of heart failure. Pulmonary oedema is about back-pressure, but raised central venous pressure and peripheral oedema is not. So what causes ankle swelling in heart failure? What follows is my understanding of what is a complicated problem. I’ll start slowly and it may take a while to get to the answer. This is the most difficult thing I’ve tried to explain so far.
First I want you to imagine a simple pump. The pump is a hollow rubbery container which works because it is made of muscle which contracts rhythmically. At each contraction the volume in the chamber is much less than when it is relaxed. For a normal left ventricle about 60% of the blood is squeezed out each time it contracts (ejection fraction).
the ventricles are made of muscle which squeezes fluid – without valves the heart would not pump in one direction
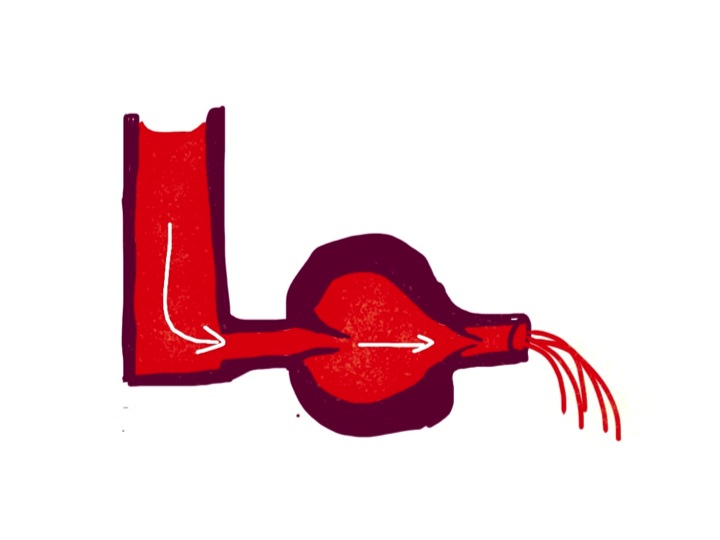
To make the blood go only in one direction, it is important to have valves at the inlet and outlet of the pump. There are three things helping to fill the pump when the muscle is relaxed. There needs to be a filling pressure, represented in the diagram below by the height of fluid in the left-hand reservoir. In the left side of the heart this is pulmonary venous pressure, in the right it is central venous pressure, most easily measured by looking at the height of blood in the neck veins. But lets first of all think about a simpler system with only one pump and one filling reservoir. Many years ago Frank and Starling discovered that if the filling pressure of a heart chamber affected how well it pumped blood out. The higher the filling pressure the stronger the contraction, making the pump work harder.
the higher the column of blood in the reservoir, the harder the heart pumps – it now has valves so the blood can only go in one direction
A lot of hollow things in our body work in a similar way. The uterus starts to contract when it gets stretched enough by the increasing size of the unborn baby. Our intestines work much harder if they are blocked, causing the wall to be stretched – this results in colic. The same is true for the gallbladder and ureters- both painfully contract when a stone causes a blockage and the muscular wall is stretched.
The second factor helping our heart chambers to fill up is suction. A turkey baster bulb will suck up fluid because the rubbery stuff it is made of wants to spring back to its normal shape. The same is true for the ventricles of the heart – they actively suck fluid in while relaxing during diastole. I am told that if you put an isolated animal heart into a bucket of oxygenated physiological saline solution it will move around like a squid – sucking liquid in and squirting it out to propel it along in the fluid. It could only do that if the ventricle actively sucks in fluid.
The third important factor in ventricular filling is the contraction of the atria. These give a bit of extra stretch to the ventricles by pumping in some more blood to help encourage them to pump it out a bit harder.
The heart pumps blood around a circuit – the circulation. It first goes into the aorta, but the vessels get progressively smaller in diameter, ending up with capillaries which have an internal diameter of less than that of a red blood cell – about 7 microns.
capillaries have just one layer of cells and are just big enough to allow a red blood cell through
Pushing blood through the large vessels is very easy, but much more difficult through the smaller vessels. There is a strange relationship between the amount of resistance to flow and the internal diameter of a pipe which was discovered by Pouseuille- resistance is inversely proportional to the fourth power of the internal diameter. In practice this means that most of the resistance to flow is in small vessels with a hole down the middle measuring less than one tenth of a millimetre – these are also known as resistance vessels. Because of Pouseuille’s law, resistance vessels can constrict or relax a very small amount to cause a large change in flow to any particular organ – depending on its requirements for oxygen.
Back to the circuit. The arteries are thick-walled, with a relatively small hole down the middle. Veins are thin walled and can accommodate more volume. So most of the blood in our circulation is in the veins. Blood is pumped into the large arteries, is squeezed through the resistance vessels, and then ends up in the veins. You will not be surprised to know that the amount of pressure in the large arteries depends both on how hard the pump is pumping – cardiac output, – and how much resistance to flow there is in the small arteries – peripheral vascular resistance. Pressure = Flow x Resistance.
One thing our bodies really care about is the pressure in our large arteries – commonly known as blood pressure. It is continually monitored by pressure-sensing devices in the aorta and carotid arteries. These baroreceptors send messages to the brain stem (medulla). If the pressure is low, the medulla sends messages via the sympathetic nervous system to the heart to make it pump harder, and to the resistance vessels to make them constrict a bit to get the pressure back to normal. The medulla also sends messages to the veins to make them constrict – we will see why in a moment.
Looking at our simple circuit, blood pressure is determined by how hard the heart is pumping, and how much resistance to flow there is in small vessels. What determines the pressure on the venous side – the filling pressure of the reservoir (on the left side in the diagram)?
blood moves around in a circuit – the pressure (height of column of blood, or central venous pressue) in the reservoir will not change if the pump is pumping fast or slowly – but the pressure on the arterial side will very much depend on how much the pump is pumping
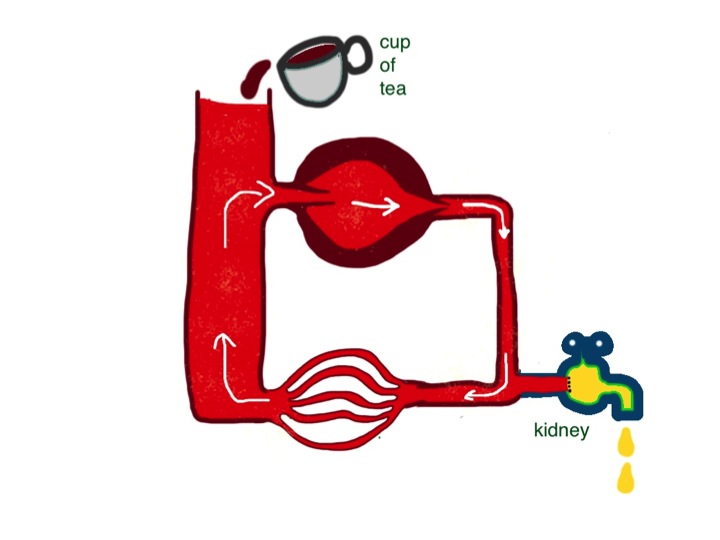
If the heart pumps less hard, or the resistance changes – I hope you can see that neither of these things will make a difference to filling pressure in the reservoir in this simple system. When students talk about “back pressure” they seem to forget that the blood goes round in a circuit. Failure of any part of the heart will not, on its own, affect filling pressure of the right side of the heart. Central venous pressure is determined by only two things: the amount of fluid in the system and how much veins are constricted. The amount of fluid in the circulation is affected by (a) how much fluid we drink every day and (b) how much we lose. Most of the fluid loss is in the form of urine (see previous post). It is the job of our kidneys to regulate the volume of blood in our circulation. Normally they do a fabulous job. If I drink a pint of beer, half an hour later I will be needing to have a pee – lots of dilute urine has been produced to get rid of all the extra water. If I eat two packets of crisps with the beer, over the next few hours my kidneys will get rid of the extra salt with no problem. Kidneys work in a timescale of hours.
central venous pressure is very much affected by the volume of blood in the circulation, which in turn depends on the balance between fluid input and output – cups of tea and daily urine volume
Venous constriction works in a timescale of seconds. If I am in a chair and about to get up and walk around, my brain stem will send messages via the sympathetic nervous system to my veins to tell them to constrict. Squeezing this complex network of floppy tubes in my legs, belly and chest will increase cardiac filling pressure and, as Frank and Starling discovered, increase the force of contraction of my heart to supply my muscles with more oxygen. Understanding of how heart failure causes problems needs an understanding of how kidneys and veins work, not just the heart.
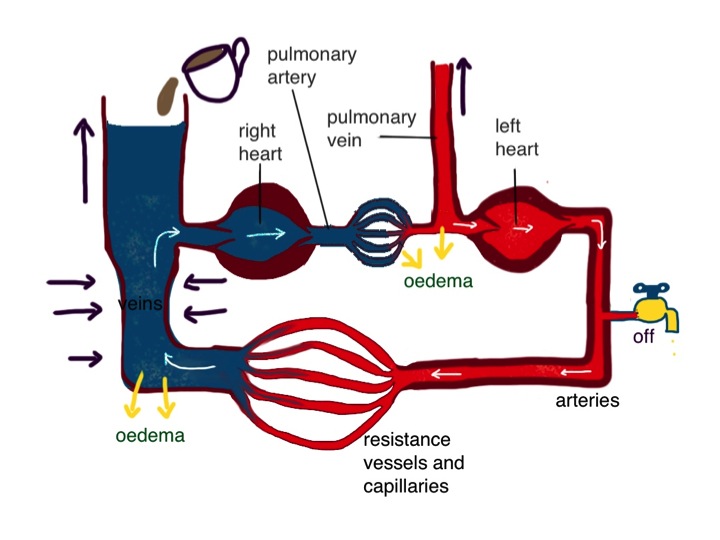
Now I am going to make the circulation model a bit more complicated. We have two pumps, not one. They are joined in series. The right heart pumps blood into the lungs. It returns to the left heart which pumps it round the rest of the body. The diagram shows the two pumps separated, but of course in real life they are part of the same organ.
this diagram shows the two sides of the heart separated – the pressure in the pulmonary vein is higher than the central venous pressure on the right side
The fact that they are arranged in series clearly could lead to problems. What if the right heart tries to pump more than the left? Why is this not normally a problem? The way the system is set up is that the right heart pumps blood through the lungs into the pulmonary veins. Here the pressure is higher than the normal 7cm water central venous pressure – in health about 16cm water. This higher pressure in the pulmonary vein is useful priming device so the left ventricle, which is bigger and stronger, can be rapidly filled and has plenty of pressure to stretch the rubbery myocardium to enable it to contract hard and generate adequate systemic arterial blood pressure. When right heart output increases, the pulmonary venous pressure will temporarily increase, and left ventricular output will in turn increase because of the Frank-Starling law, and subsequent reduction of pulmonary venous pressure to normal. The end result is that the output of the left side of the heart matches that of the right, and pulmonary venous pressure has not changed much – all sorted. This does mean, if you think about it, that the cardiac output is being controlled by the right heart – it pumps what it wants and the left heart has to follow suit. It is not always the case that the bigger, stronger partner is in control. John has found that out recently.
What controls right heart output? Well the brain stem has a big say, by constricting veins and increasing filling pressure and by sending messages to the heart via sympathetic nerves. But the kidneys are also really important – by altering fluid balance they can control filling pressure. Kidneys are involved in turning the dials and can really mess things up when they get it wrong, as we shall see.
Lets get back to John. His left ventricle was damaged by a sizeable myocardial infarction a few years ago (see chest pain and horsemeat lasagne below). Soon after had an echocardiogram which showed that the part of his left ventricle was not contracting as well as it should – the overall ventricular ejection fraction was found to be 44%. It was probably about 60% before. The rest of his left ventricle which was not damaged has to work harder to maintain his cardiac output and blood pressure. Heart muscle is different from ordinary skeletal muscle – the sort which makes our arms and legs work. Skeletal muscle responds very well to being worked hard. It grows bigger and stronger to more exercise we take. The heart muscle does this to an extent, but eventually it seems to give up and stop working so well. I don’t think anyone quite understands why this is. If experimental animals are infused with isoprenaline, a drug which makes the heart beat strongly by stimulating heart muscle cells in a similar way to noradrenaline released by sympathetic nerves, their hearts will fail in about 2 weeks. Beta blockers stop this sympathetic stimulation, and patients with heart failure live longer if they take them regularly.
Poor cardiac output stimulates the kidneys to produce renin. This is an enzyme which generates angiotensin I. Conversion of angiotensin I to angiotensin II on the surface of blood vessels helps restore blood pressure but also has a bad effect on heart muscle. It seems to make the muscle cells produce damaging free-radicals and oxidising agents which cause cardiac muscle death. ACE inhibitors prevent the formation of angiotensin II and protect the heart. But despite these drugs, once the ventricle is badly damaged, often the remaining heart muscle starts to fail after a period of time. Clearly in John’s case, another small myocardial infarction may have tipped him into symptomatic heart failure, despite the statins, omega-3 (fish oil), aspirin, and lack of steak and kidney pudding.
What happens when the left ventricle fails to pump properly? The right heart output is determined by central filling pressure and sympathetic activity. If it pumps blood through the lungs to the pulmonary veins, and if the left ventricle is not working, it will cause the pulmonary venous pressure to rise – lets say from the normal 12cm water to 20cm water. This rise in pressure will force the damaged left ventricle to pump harder until it can remove all the blood from the lungs that is being delivered. The right heart will not be too pleased with this- it is trying to pump what it thinks is the right amount of blood but is having a problem. As the pulmonary venous pressure rises, assuming the resistance to flow through the lungs does not change, the pressure in the pulmonary artery rises by the same amount as the rise in pulmonary venous pressure. The right ventricle will have to do more work to pump out blood into the lungs. Now, the left heart normally does not mind pumping at high pressure and doing lots of work – that’s what it is designed for. The right heart is just no good at it – it’s not lazy but it just does not have the muscle. (I put that bit in in case my wife decides to read this).
when the left ventricle fails it cannot pump blood out of the lungs and pulmonary venous pressure rises – reduced arterial pressure is sensed by baroreceptors and the kidney – reflex venoconstriction and reduction in urine output lead to a rise in central venous pressure which makes the right heart pump more blood into the lungs – increasing pulmonary oedema – fluid retention by the kidneys is the cause of peripheral oedema, not “back pressure”
So, as a result the right heart can pump out less blood and we are left with a new balance – lower cardiac output and higher pulmonary venous pressure. The right heart is under strain because it is having to pump against higher pressure and the left heart is under strain because its filling pressure is higher than normal, making the undamaged parts also work harder than normal.
That’s fine until the kidneys and the brain get involved. They were doing a good job when John was healthy but in a crisis they make some bad management decisions. When cardiac output drops, blood pressure drops in proportion. Baroreceptors sense this and let the medulla know “Houston, we have a problem”. Increased sympathetic supply to small blood vessels is a good idea. Noradrenaline is released from nerves on the surface of blood vessels acting on alpha 1 receptors in the smooth muscle cell membrane. This causes the vessel to contract, making the holes down the middle smaller. Peripheral resistance increases and so does blood pressure. That was a good decision. There are similar sympathetic nerves which supply the heart. They are activated and the nerves again release noradrenaline, this time acting on beta receptors. The effect is to increase the rate and force of contraction of the heart, increasing cardiac output and blood pressure. Fine in theory, but this increase in work rate will increase cardiac oxygen consumption. John’s heart has a problem with getting enough oxygen because his coronary arteries have been ruined by too many steak and kidney puddings. Sympathetic stimulation can do more harm than good –that’s when reading the theory book goes wrong – a bad decision. And that is why we us beta blockers in patients with coronary artery disease and give them in patients who have had a myocardial infarction.
Another bad decision is to send messages to the veins to constrict. Increasing right heart filling pressure seems like a good idea. The right heart responds by pumping more blood into the lungs, not aware that the left heart is having a problem. If the pulmonary venous pressure goes above 25cm water there are serious problems. Fluid starts leaking out of the circulation in the lungs and causes pulmonary oedema. The fluid causes swelling of the gas-exchange membrane which allows oxygen to pass from the lungs to the blood, and for carbon dioxide to get out. Increased carbon dioxide concentration and reduced oxygen in the bloodstream are sensed by chemoreceptors in the carotid artery and the medulla (the respiratory centre which controls breathing is a close neighbour to the vasomotor centre which controls blood pressure). Houston panics. John (and his wife) also panics – he is fighting for breath. Houston is in panic mode – not only are its baroreceptors and chemoreceptors telling it that things are going wrong – messages are coming down from the White House – the cerebral cortex.
“More sympathetic stimulation” says Houston. Another bad decision. The increased sympathetic increases venous constriction and right heart filling. We are in a bad visious cycle, throwing petrol onto the flames. Just when Houston was beginning to give up Superwoman arrives. The nice ambulance woman has been trained not to panic – she’s seen it all before. It took her about 2 seconds to realise John had acute pulmonary oedema. She sat him up, gave him oxygen and got him to open his mouth and sprayed nitroglycerin under his tongue. Don’t worry dear- you’ll be fine in a moment. And he was. Reassurance works wonders in acute pulmonary oedema by reducing panic. Nitroglycerin or glyceryl trinitrate as we call it in the UK also works very well by selectively dilating veins, having less effect on the small resistance arteries. This reverses the bad vicious cycle, reducing right heart filling pressure, reducing right ventricular output and reducing pulmonary oedema, which reduces carbon dioxide levels in the blood which makes Houston much happier “was that a bird or a plane?”
I know of no evidence to support this, but I feel pretty sure the reason opiates like morphine work to reduce pulmonary oedema is to supress the respiratory centre and reduce central sympathetic output. Opiates certainly don’t work directly on veins to cause vasodilatation.
The problem with the medulla and normal cardiovascular reflexes is that they were designed to get us out of trouble when blood pressure falls from volume loss from trauma, diarrhoea or sepsis. Blood pressure drop from left ventricular failure is not in the manual. Primitive humans did not eat steak and kidney puddings.
The kidney is no better when it comes to heart failure. It knows there is a problem with cardiac output because it can sense it not getting enough blood. It makes the wrong assumption that this is due to circulating volume loss. The kidney’s response to low cardiac output is to stop producing urine. John is still drinking cups of tea and eating salty food, but he is not excreting it. The salt and water has to go somewhere – it increases the volume of blood in the circulation and increases right heart output, predisposing towards pulmonary oedema. The increase in blood volume can only go so far before it leaks out of the circulation and then equilibrates with extra-vascular interstitial fluid to produce ankle oedema. The only way to sort this out is to give diuretics like furosemide. This will force the reluctant kidneys to produce more urine and get the input/output balance right.
Many patients are fearful of taking daily furosemide, thinking that it will make them pass lots of urine all the time and make their lives difficult. Certainly, after the first few doses, daily urine volume increases, but it does not take much thinking about to realise that urine volume will soon settle down to be the same as the volume of fluid drunk each day. It can alter the pattern of urine output, so that for six hours after taking furosemide the volume is higher than normal, but for the next eighteen hours it is less. It might mean that patients need to get up at night to pass urine less often. Furosemide used to be called Lasix, because its effect lasts six hours.
I have explained why patients with heart failure benefit from nitroglycerin, morphine, beta blockers and furosemide. What about ACE inhibitors. As well as producing of free radicals in the heart, angiotensin II has an effect on veins. It does not directly constrict them, but enhances the effect of sympathetic activation originating in the medulla. That means that if the circulating levels of angiotensin II are very high, which is the case in heart failure, normal exercise will result in an exaggerated constriction of veins, an excessive rise in central venous filling pressure and too much blood being pumped into the lungs which the damaged left ventricle can’t handle. This will result in breathlessness. The main benefit patients with heart failure notice when they start ACE inhibitors is a reduction of breathlessness on exertion. The reduction of free radical production is a separate long-term benefit which will keep John alive longer.
So why do students (and doctors) talk about right heart failure as the cause of fluid retention and raised venous pressure when the damage is to the left side of the heart? I think the most likely reason is that in patients with true right heart failure, secondary to severe lung disease or pericardial tamponade, oedema can become really impressive. But again, the cause is not “back pressure”. If the right heart fails to pump properly, say the cardiac output drops from 5 litres/min to 2 litres/min, then the left heart can only pump out 2 litres/min. This means that the kidney again mistakenly goes into shutdown mode and fluid accumulates. The difference here is that pulmonary oedema is not going to be a problem, as the left heart is working normally, keeping pulmonary venous pressure normal. Under these circumstances patients sometimes present with gross oedema in their legs and fluid in their abdominal cavity, without presenting earlier with breathlessness. Thus historically, gross oedema is associated with right heart failure. Unless you can think of a better reason.
I can’t finish discussion of heart failure without mentioning preload and afterload. Anyone who mentions these terms when talking about humans, in general, is using the terms inappropriately. Preload and afterload were terms invented, I think, by Braunwald and Epstein. They were two brilliant cardiological physiologists who were interested in how filling pressure and arterial pressure affected cardiac ventricular function. They devised very clever experiments on isolated rat heart ventricular strips in organ baths. They wanted to look at the effect of increased filling pressure – so they put a spring in the organ bath to produce more tension and looked at how that affected contraction – they called it preload but never meant it to be anything but a proxy for filling pressure. Similarly, they thought “how can we model how the ventricle contracts against higher arterial pressure?” They cleverly added weights to be pulled up by the contracting ventricular strip – to make the ventricle do work. They called this afterload, but again did not mean it to be used to describe the in-vivo situation. If I ask one of the junior doctors “what is that patient’s preload?” there is no answer. They can tell me that the neck veins are distended. If I ask “what is that patient’s afterload?” they will rightly look at me strangely. But of course they can tell me what his blood pressure is.
The food link this week is haggis. This is traditionally made from heart, lungs and kidney. Particularly appropriate for this discussion on heart failure. I like haggis roasted, not boiled. When roasted the skin, made from a sheep’s stomach, goes all brown and crispy. You must serve it with neeps (bashed swede, or rutabaga in the US) and tatties (mashed potato). Gravy is good but not necessary if you have a glass of whisky with it. Unfortunately I do suspect it is not much better than steak and kidney pudding for my coronary arteries as it also contains suet – the most saturated fat available. But it does have oatmeal.